
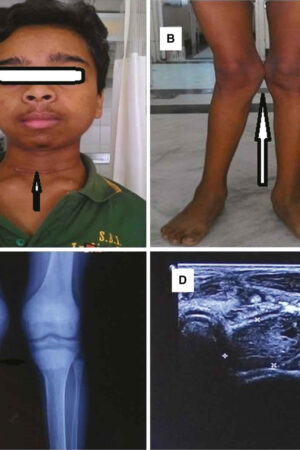

Pseudoachondroplasia
Pseudoachondroplasia is an inherited disorder of bone growth, a type of short-limbed dwarfism. Pseudoachondroplasia was once thought to be related to another disorder of bone growth called achondroplasia, but without that disorder’s characteristic facial features. More research has demonstrated that pseudoachondroplasia is a separate disorder.
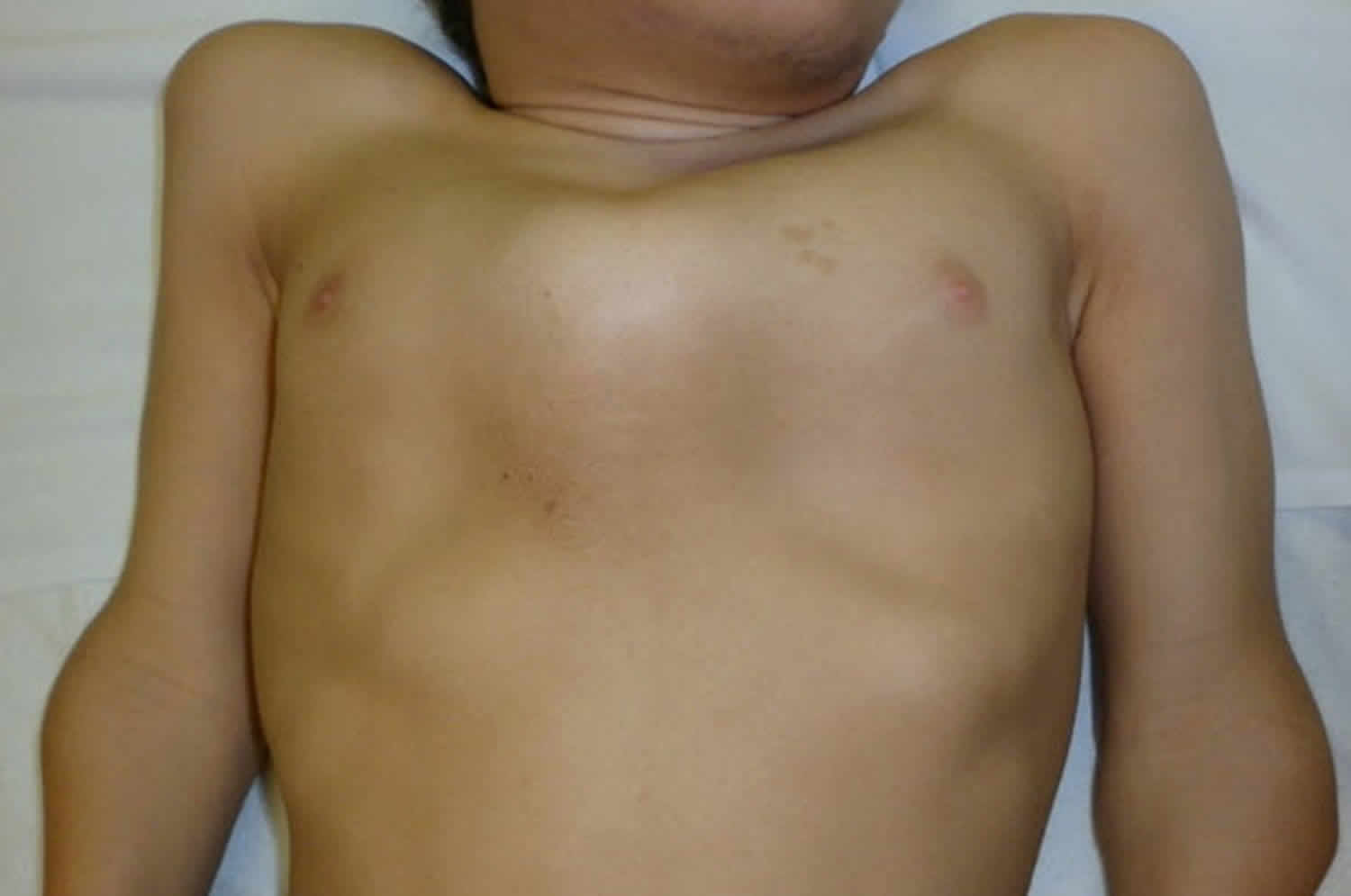
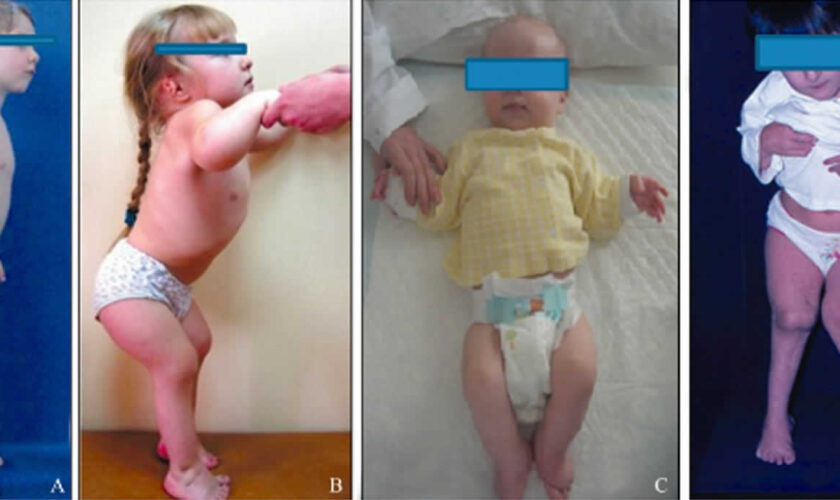
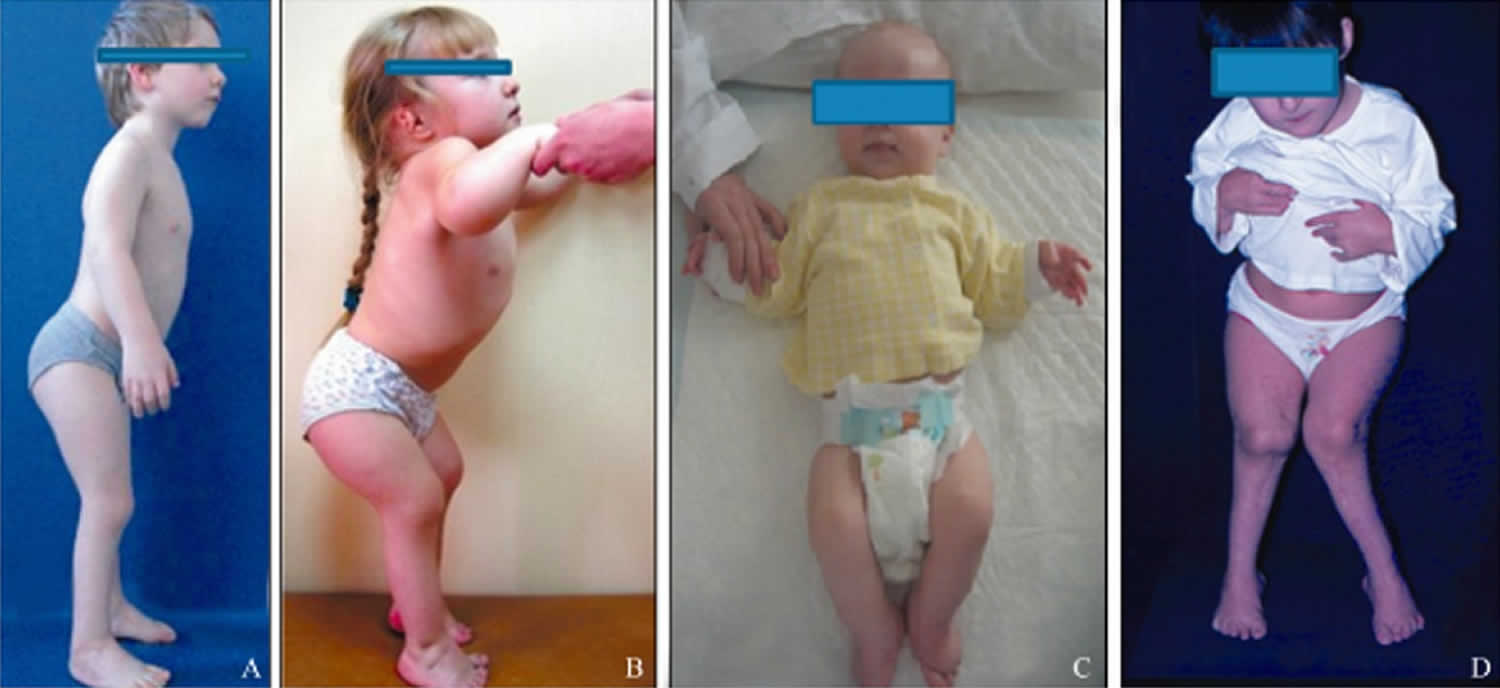
All people with pseudoachondroplasia have short stature. The average height of adult males with this condition is 120 centimeters (3 feet, 11 inches), and the average height of adult females is 116 centimeters (3 feet, 9 inches). Individuals with pseudoachondroplasia are not unusually short at birth; by the age of two, their growth rate falls below the standard growth curve.
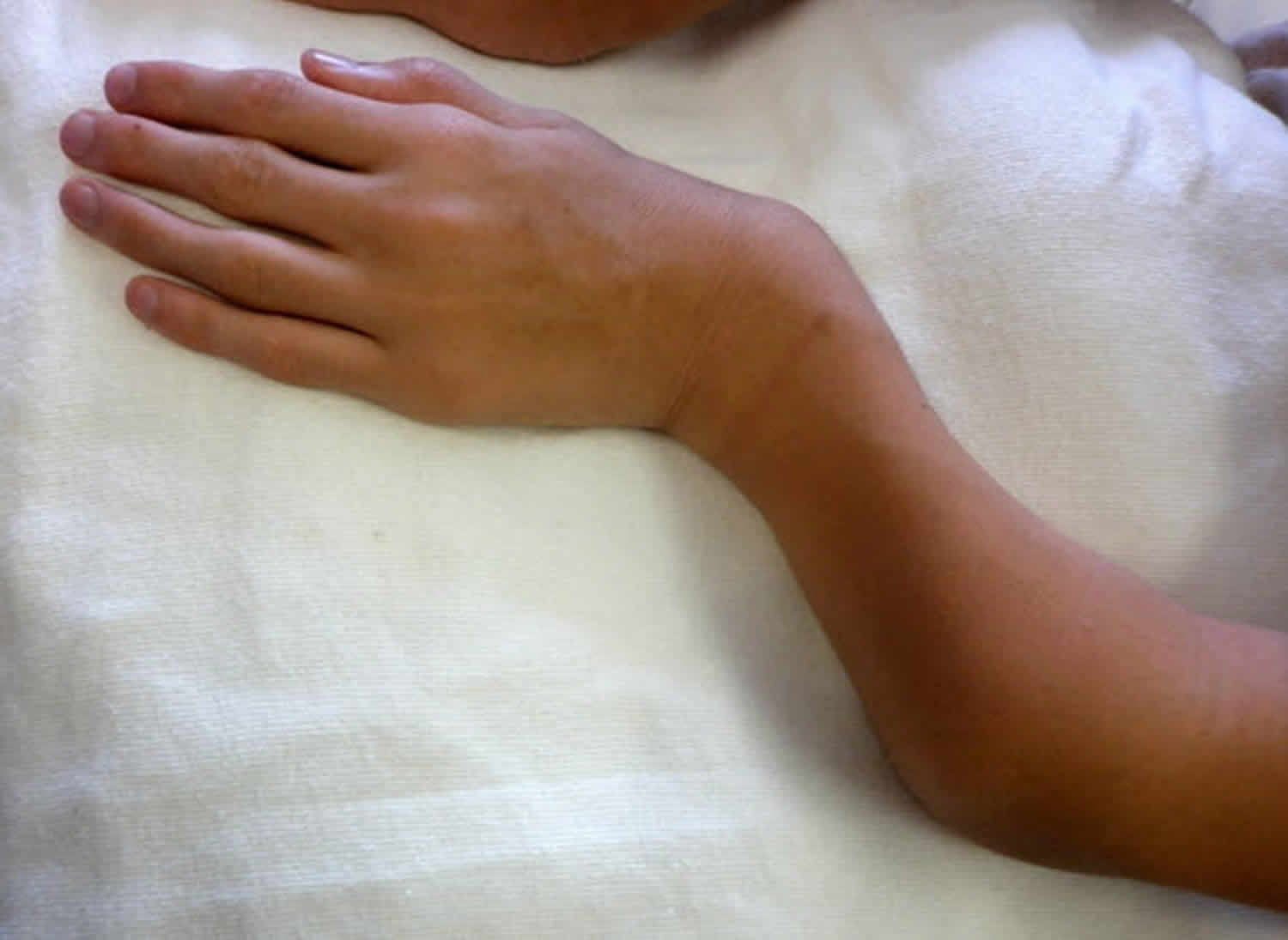
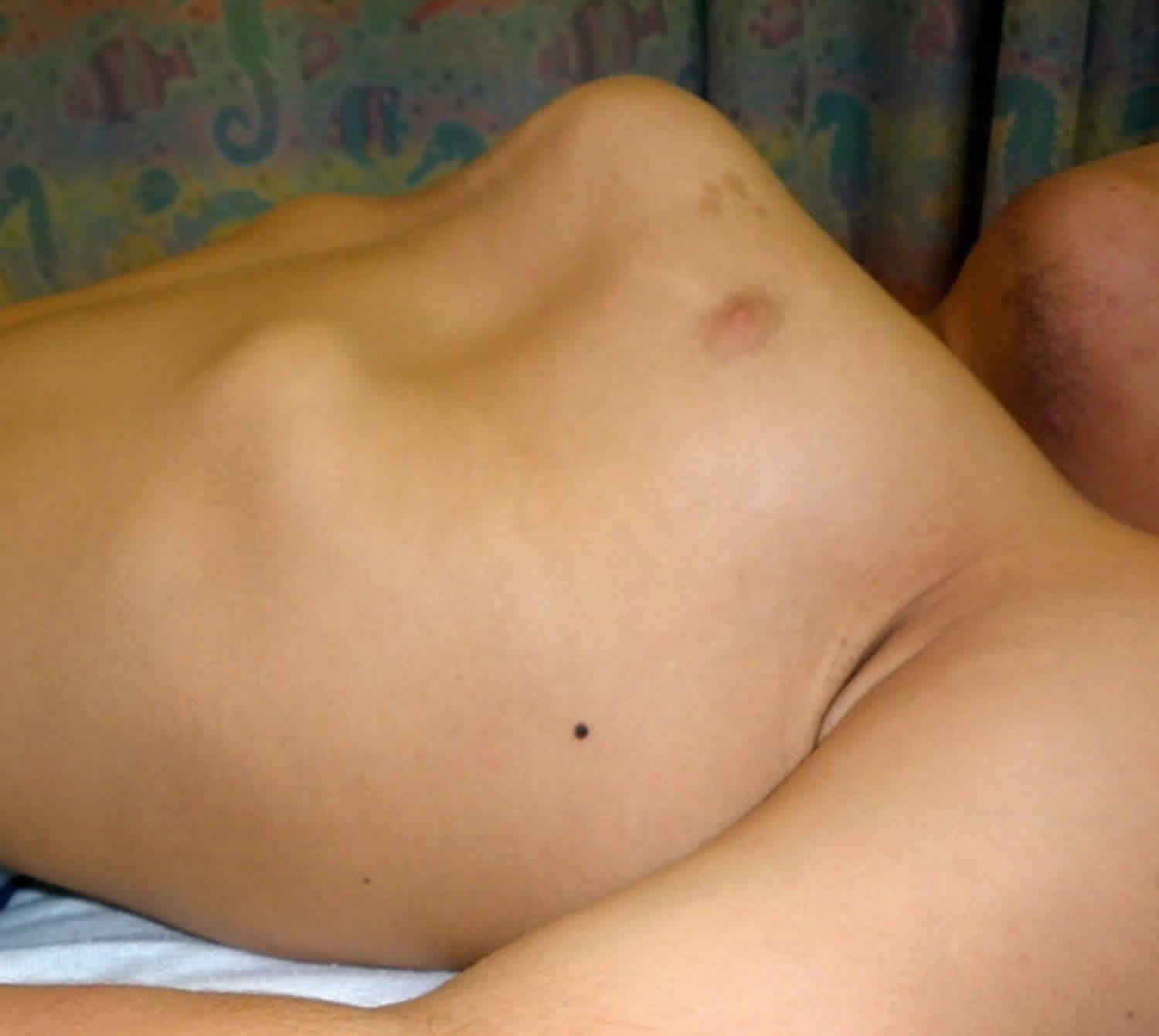
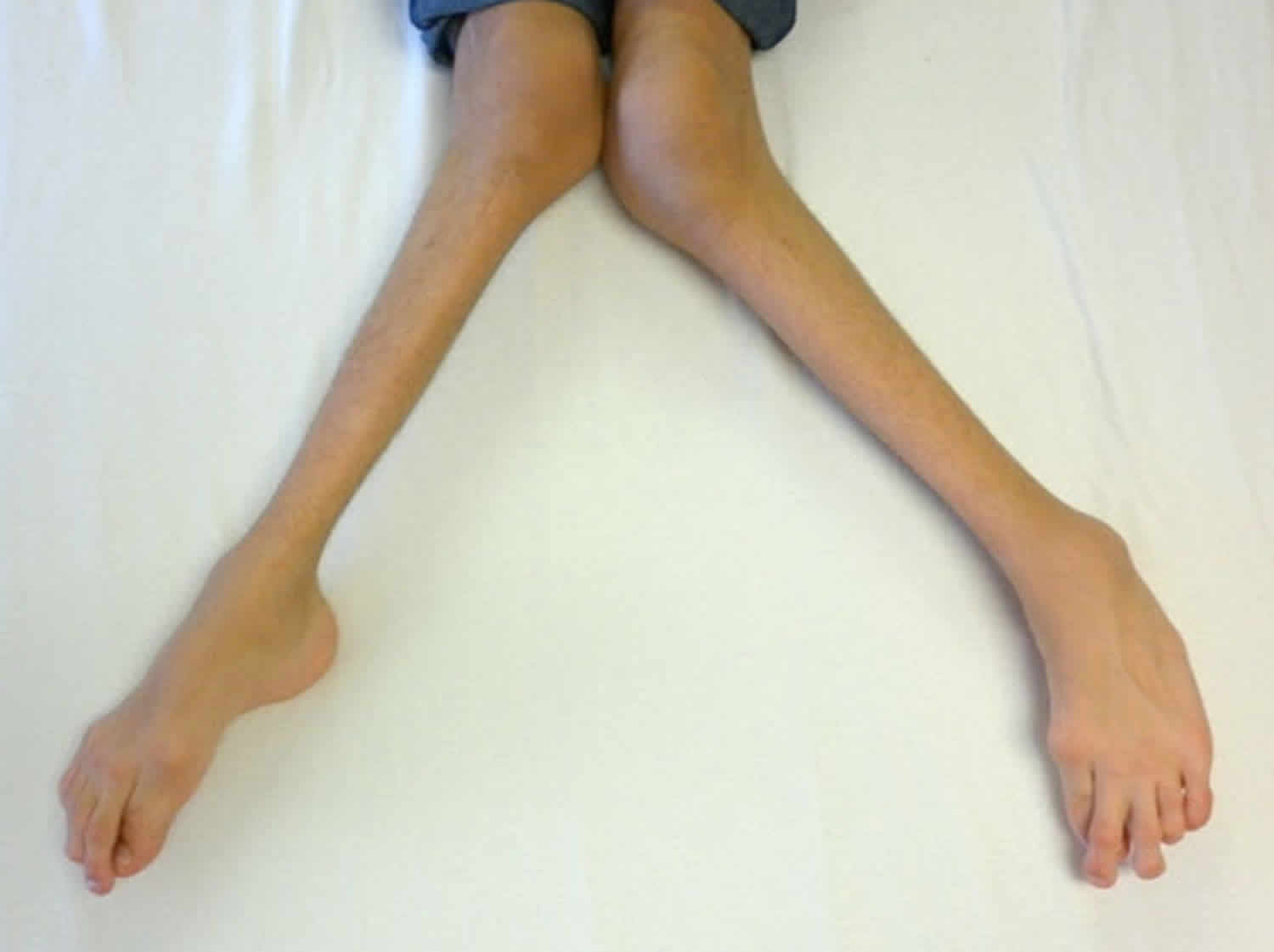
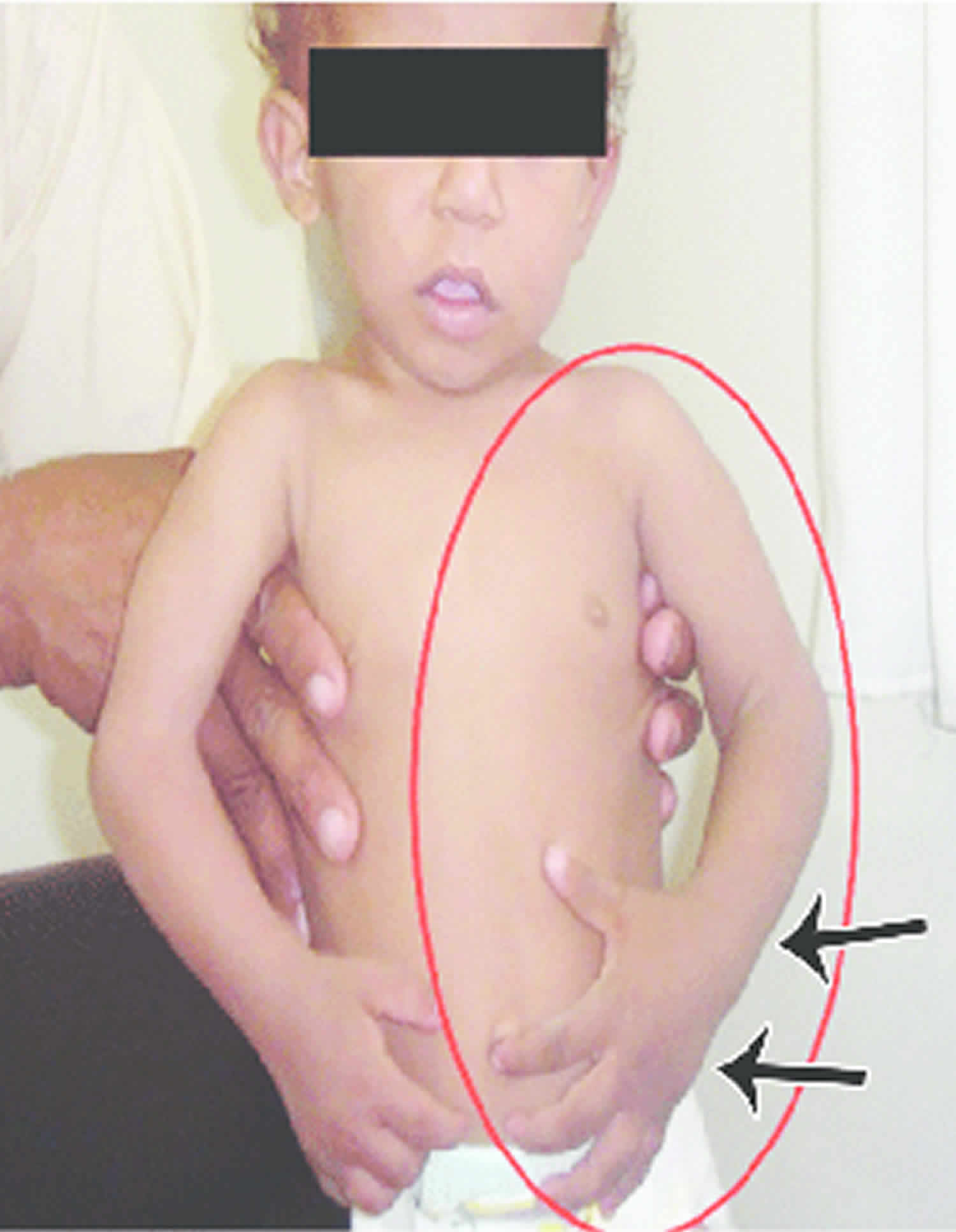
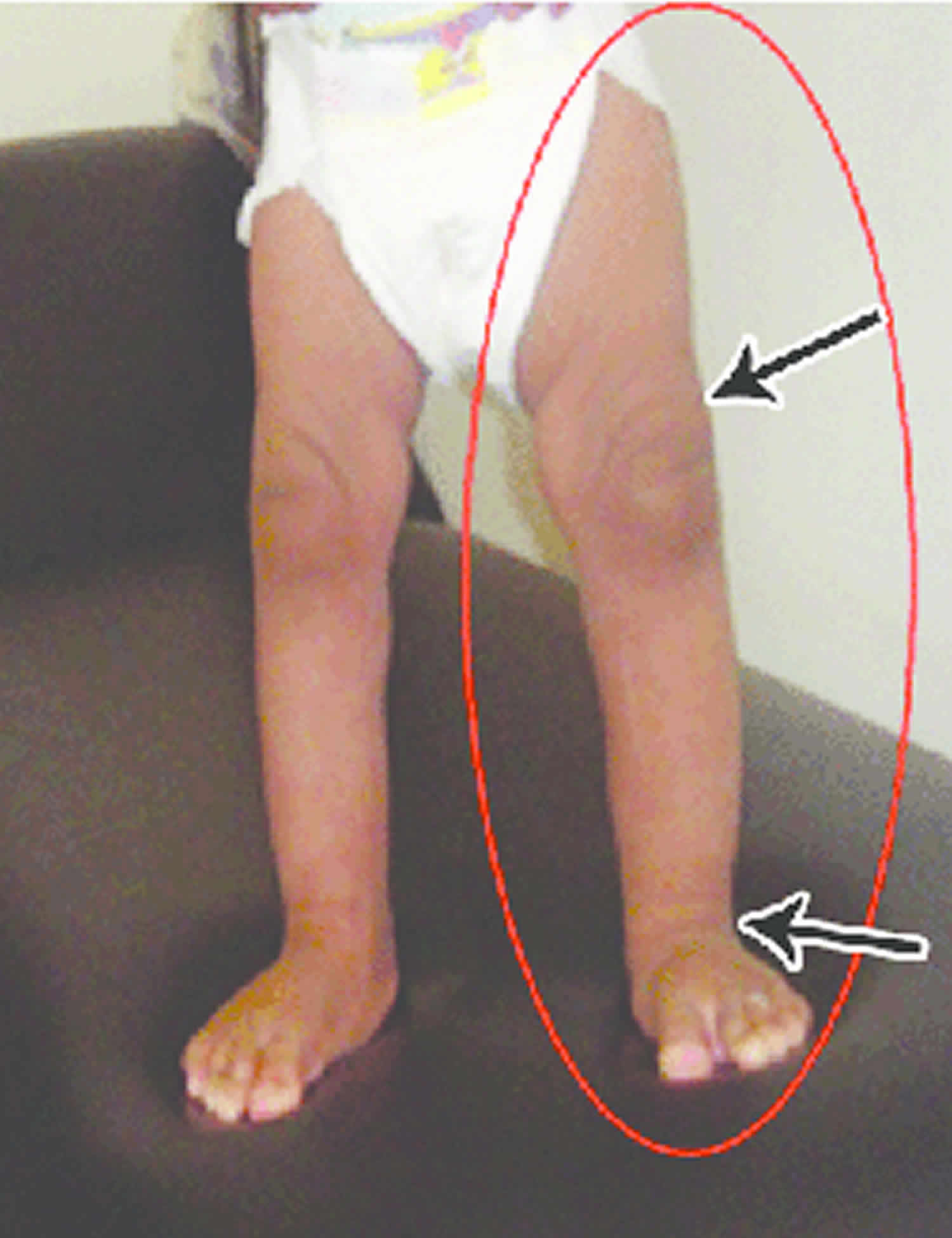
Other characteristic features of pseudoachondroplasia include short arms and legs; a waddling walk; joint pain in childhood that progresses to a joint disease known as osteoarthritis; an unusually large range of joint movement (hyperextensibility) in the hands, knees, and ankles; and a limited range of motion at the elbows and hips. Some people with pseudoachondroplasia have legs that turn outward or inward (valgus or varus deformity). Sometimes, one leg turns outward and the other inward, which is called windswept deformity. Some affected individuals have a spine that curves to the side (scoliosis) or an abnormally curved lower back (lordosis). People with pseudoachondroplasia have normal facial features, head size, and intelligence.
Common characteristics of pseudoachondroplasia:
- Can affect boys and girls equally
- Diagnosis may not occur until around 2 years of age, when physical characteristics maybe apparent.
- Shortened Limbs
- Short stubby fingers
- Waddling walk
- Joint pain (with age)
- Large range of joint movement (hyperextensibility) in hands, knees and ankles
- Limited range of motion in the elbows, and hips
- Some people have legs that turn outwards (valgus) or inwards (varus). Occasionally have one leg turning in, the other turning out (windswept)
- Sometimes have curved spine (scoliosis)
- ‘Normal’ facial features
- Life expectancy is not affected
- Intelligence is not affected
Pseudoachondroplasia is caused by a mutation in the cartilage oligomeric matrix protein (COMP) gene and is transmitted in an autosomal dominant pattern. Thirty percent of cases are familial with an affected parent transmitting the condition, while 70% occur as a random, new (de novo) mutation in COMP with no previous family history.
The exact birth prevalence pf pseudoachondroplasia is unknown, but estimated to be 1 in 30,000-100,000. Males and females are equally affected.
Treatment for pseudoachondroplasia varies because the condition affects several body systems, and each child’s case is different. Some children will only require careful monitoring. Others will need, non-surgical or surgical treatments to address specific aspects of their condition.
Many children with pseudoachondroplasia are also diagnosed with a variety of orthopaedic conditions including: scoliosis, hip pain, joint stiffness and limb shortening. In most cases, these conditions only become evident – or problematic – as your child grows. Depending on your child’s needs, orthopaedic specialists will treat your child.
Every child’s condition is different, so treatment is determined on a case-by-case basis. For example, if your child has scoliosis, spine specialists will consider the severity of the curve, where it occurs in the spine, and your child’s age and stage of growth, before determining the best course of action. Treatment may include non-surgical options such as bracing and physical therapy, or surgical options such as spinal fusion or implanting growing rods to stabilize your child’s spine as he continues to grow.
For other effects of pseudoachondroplasia, in general, treatment may include:
- Bracing and/or surgery for scoliosis
- Medication or pain relievers for joint pain
- Bracing and/or surgery for knee and lower-leg deformities
- Bracing and/or surgery to treat hip pain
- Physical therapy to help your child remain limber
Pseudoachondroplasia causes
Mutations in the COMP (cartilage oligomeric matrix protein) gene cause pseudoachondroplasia. The COMP gene provides instructions for making a protein that is essential for the normal development of cartilage and for its conversion to bone. Cartilage is a tough, flexible tissue that makes up much of the skeleton during early development. Most cartilage is later converted to bone, except for the cartilage that continues to cover and protect the ends of bones and is present in the nose and external ears.
The COMP protein is normally found in the spaces between cartilage-forming cells called chondrocytes, where it interacts with other proteins. COMP gene mutations result in the production of an abnormal COMP protein that cannot be transported out of the cell. The abnormal protein builds up inside the chondrocyte and ultimately leads to early cell death. Early death of the chondrocytes prevents normal bone growth and causes the short stature and bone abnormalities seen in pseudoachondroplasia.
Pseudoachondroplasia inheritance pattern
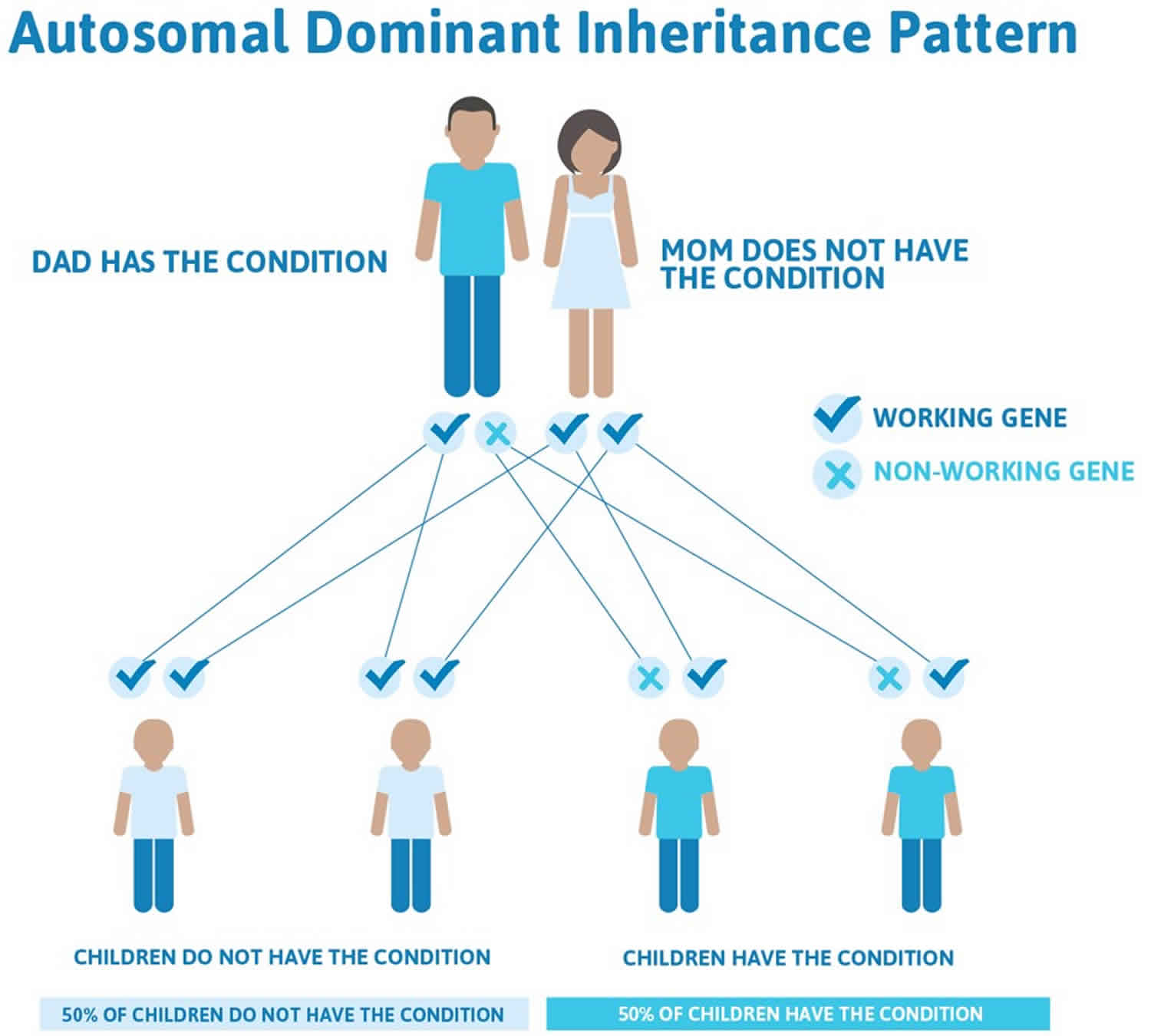
Pseudoachondroplasia is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In some cases, an affected person inherits the mutation from one affected parent. Most cases (70% of cases) result from new mutations in the gene called new (sporadic or de novo) mutation and occur in people with no history of the disorder in their family. In sporadic or de novo mutation case, pseudoachondroplasia is usually not inherited from or “carried” by a unaffected parent. However, once the mutation has occurred, it is transmitted by dominant inheritance from either an affected mother or father to their child. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disorder. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the child.
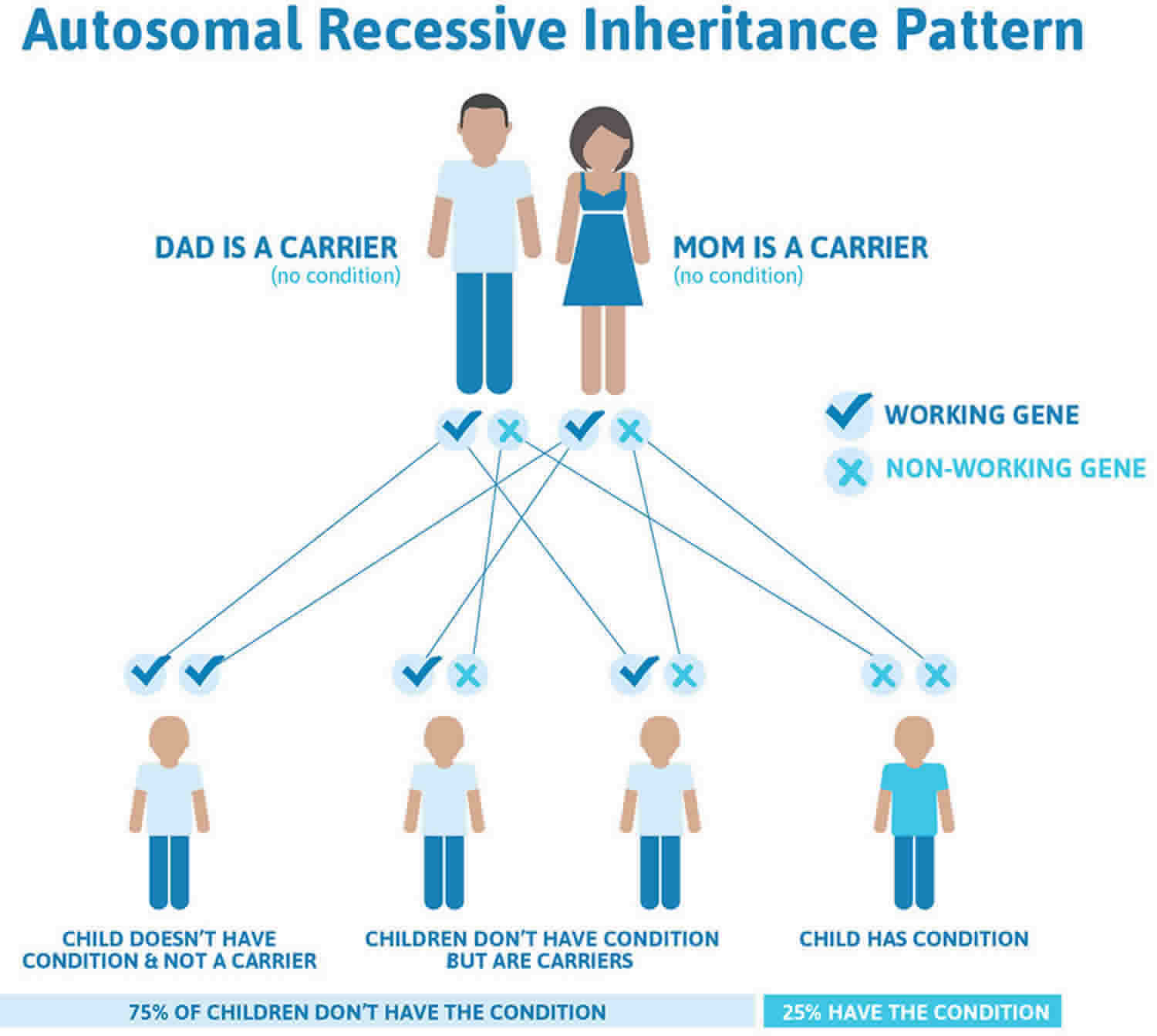
A few families showed recurrence in what appeared to be autosomal recessive inheritance. Mutational analysis revealed that these cases were the result of parental germline mosaicism for a COMP mutation. As a result, one or more of the parent’s children may inherit the germ cell gene COMP mutation, leading to pseudoachondroplasia, while the parent does not have this disorder because the mutation is not present in sufficient number of body cells. The likelihood of a parent passing on a mosaic germline mutation to a child depends upon the percentage of the parent’s germ cells that have the mutation versus the percentage that do not. There is no test for germline mutation prior to pregnancy. Testing during a pregnancy for familial cases with a known mutation is available and should be discussed with a genetic specialist.
Figure 1. Pseudoachondroplasia autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Pseudoachondroplasia symptoms
Signs of pseudoachondroplasia can vary from child to child, but may include:
- Short arms and legs
- Delay in crawling and walking
- Waddling walk
- Joint pain at an early age
- Limited range of motion at the elbows and hips
- Abnormally large range of joint movement (hyperextendibility) in the hands, knees and ankles
- Knee deformities, such as bow-legs (knees that point outward) or knock-knees (knees that point inward)
- Curvature of the spine, including scoliosis or lordosis, an inward curvature of the cervical (top) and lumbar (middle) portions of the spine
- ‘Normal’ facial features
Pseudoachondroplasia shows variable expression with the severity varying within and between families. Infants with pseudoachondroplasia have normal birth parameters and cannot be distinguished from unaffected newborns since birth length and weight are normal. Generally, the first sign is diminished linear growth starting between 9 to 12 months with length and eventually height falling approximately two years behind the standard growth curve. Disproportionate short stature becomes more apparent with age. Affected children usually begin to walk between 12- 18 months but the gait is abnormal and described as ‘waddling’ reflecting underlying skeletal abnormalities involving the hips. The face is attractive and has been described as angular. Disproportionate shortening of the arms and legs become apparent between 3-5 years of age. The hands and feet are short with the fingers and toes showing dramatic shortening and laxity. All the joints exhibit extreme laxity except the elbows which may have limited extension. The joint laxity at the knees contributes to the lower extremity deformities that include bowing (genu varum) or knock knee (genu valgum) deformities. Sometimes bowing can occur in one leg and a knock knee deformity in the other. Surgical correction is generally required but should be delayed to get maximum sustainable correction.
Spinal abnormalities are common and include: 1) scoliosis or S-shaped spinal curve, 2) exaggerated lumbar lordosis, which is an abnormal inward curvature of the lower portion of the spine and 3) kyphosis, which is abnormal front-to-back (or outward) curvature of the spine so that the spine is abnormally rounded at the top. Underdevelopment (hypoplasia) of the small, tooth-like projection (odontoid) at the top of the spine occurs infrequently. Odontoid hypoplasia causes instability in the neck region (cervical instability), which increases the risk of spinal injury (cervical myelopathy). This complication often requires surgical fusion of the upper spine.
Pain, a common and universal complaint, starts in early childhood and is exacerbated by exercise. Activities that stress the joints should be avoided. This includes all contacts sports and trampoline. Early joint pain may reflect an inflammatory process related to the underlying chondrocyte pathology. Osteoarthritis in early adulthood is a universal finding usually developing into chronic joint pain (arthralgia). The hips, ankles, shoulder, elbows and wrists are particularly affected. Degenerative joint disease is progressive and ultimately may require surgery starting with hip replacement followed by other joint replacements. Symptomatic treatment with anti-inflammatory medications is used for pain management with varying degrees of success.
Final adult height on average is 3’8” (116 cm) in women and 3’9” for men (120 cm) but this can vary as some individuals may attain a height of 4’10”. Intelligence and life expectancy are unaffected and most individuals raise families and lead productive, active and full lives.
Pseudoachondroplasia diagnosis
The diagnosis of pseudoachondroplasia is based upon identification of characteristic clinical and radiographic findings, detailed patient history, and genetic testing. The diagnosis is rarely made at birth because short stature is not present. The distinctive features develop over time, and this sets it apart from other short stature conditions.
Clinical testing and workup
A complete set of x-rays (radiographs) can help to establish a diagnosis by revealing abnormal growth centers (epiphyses) and other characteristic skeletal findings. The diagnosis is made clinically and by reviewing the radiographs. More advanced imaging techniques such as magnetic resonance imaging (MRI) and computed tomography (CT) scans can be used later to assess skeletal health, particular in advance of surgery to correct skeletal malformations. An MRI uses a magnetic field and radio waves to produce cross-sectional images of particular organs and bodily tissues. During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of certain tissue structures.
Genetic counseling helps families understand the genetics and natural history of pseudoachondroplasia as well as providing psychosocial support. Sequencing of the COMP gene confirms the diagnosis and is commercially available. Prenatal diagnosis for pregnancies at increased risk for pseudoachondroplasia is accomplished by chorionic villus sampling or amniocentesis if the COMP gene mutation has been identified in an affected family member.
Pseudoachondroplasia treatment
Treatments are directed toward the specific symptoms as they become apparent and usually require the coordinated efforts of a team of specialists. The team includes geneticist, pediatrician, specialists in treating skeletal disorders (orthopedic surgeons), neurologists, physical and occupational therapists and other healthcare professionals who will systematically and comprehensively plan needed treatments.
Specific therapeutic procedures and interventions may vary, depending upon numerous factors, such as disease severity; the presence or absence of painful symptoms; an individual’s age and general health; and/or other elements. Decisions concerning the use of particular drug regimens and/or other treatments should be made by physicians and other members of the health care team in careful consultation with the patient based upon the specifics of his or her case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
Pain medications may be beneficial in treating pain associated with joint disease. Physical therapy, which can improve joint motion and avoid muscle degeneration (atrophy), is beneficial.
In some patients, surgery may be necessary to achieve better positioning and to increase the range of motion in certain joints. Surgery may be necessary to treat malformation of the hips and, in some individuals, total hip replacement surgery (total hip arthroplasty) may be necessary. Surgical procedures may be recommended to treat abnormalities of the knees and lower legs. Osteotomy, a surgical procedure in which bone is cut to change the alignment, is common in pseudoachondroplasia to treat improper alignment of bones of the lower legs.
In some children, spinal abnormalities may require surgical intervention. Abnormal curvature of the spine, e.g. scoliosis, usually does not require surgery, but in severe cases, surgery has been effective. More serious spinal problems such as cervical instability may require spinal fusion.
Follow-up care
Your child with pseudoachondroplasia should be monitored by an orthopaedic physician throughout his development, into adulthood.
Doctors will watch for degenerative joint disease and neurological problems (such as weakness in an arm or leg), and assess lower-limb alignment and joint pain. It is not uncommon for children with pseudoachondroplasia to have legs that are slightly different lengths, which can affect the way the child walks (gait) in the short-term, and can affect hip function in adulthood.
If your child had spine, hip or leg surgery, he or she will need to see the orthopaedic surgeon about one to two weeks after surgery, then again at three and six months post-surgery. After that, annual monitoring by trained clinicians is strongly encouraged to ensure any problems are spotted and treated as soon as possible.
Additionally, physicians may recommend your child see several specialists because other body systems may be affected by pseudoachondroplasia.
For example, your child may see:
- An orthopaedist for any bone-, muscle- and joint-related issues
- Physical therapists and occupational therapists to expand your child’s physical dexterity and skill
- A neurologist or neuromuscular specialist to address any nerve or muscle weakness
- A psychologist or social worker to address any body-image and related mental health issues
During follow-up visits, X-rays and other diagnostic testing may be done. The goal of continued monitoring is to help spot any irregularities in growth or development and to address health issues as they develop.
Pseudoachondroplasia prognosis
Children with pseudochondroplasia can lead relatively normal lives. They have normal intelligence and an average life span.
Ongoing medical monitoring is important for people with pseudoachondroplasia. About half will eventually require hip replacement, and some may develop arthritis or further spine problems.
If you have questions about how your child’s condition and any related health issues may affect your child’s prognosis or long-term goals, talk to your child’s healthcare provider.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}