Contents
Pulmonary hypoplasia
Pulmonary hypoplasia also called lung hypoplasia refers to deficient or incomplete development of parts of the lung, which can be unilateral or bilateral 1). Pulmonary hypoplasia is characterized by small, underdeveloped lungs that can affect not only breathing but also heart function, ability to feed, hearing and overall development. Some children with pulmonary hypoplasia develop a related condition known as pulmonary hypertension, which causes high blood pressure in the arteries of the lungs (the pulmonary arteries). Over time, this pressure causes the pulmonary arteries to narrow, making the right side of the heart work harder as it forces blood through the narrowed arteries.
The true prevalence of pulmonary hypoplasia is unknown. The reported incidence is between 9 to 11 per 100,00 live birth which is an underestimation, as infants with lesser degrees of hypoplasia likely survive in the neonatal period 2). Incidence also varies by cause. In cases of premature rupture of membranes at 15-28 weeks gestation, the reported prevalence of pulmonary hypoplasia ranges from 9 to 28% 3). Most cases are secondary to congenital anomalies (such as congenital diaphragmatic hernia and cystic adenomatous malformations) or complications related to pregnancy that inhibit lung development. These include, but are not limited to, renal and urinary tract anomalies, amniotic fluid aberrations, diaphragmatic hernia, hydrops fetalis, skeletal and neuromuscular disease and conditions like pleural effusions, chylothorax and intrathoracic masses that cause compression of the fetal thorax 4).
Pulmonary hypoplasia may be primary or secondary. Primary pulmonary hypoplasia is extremely rare and routinely lethal. The severity of the lesion in secondary pulmonary hypoplasia depends on the timing of the insult in relation to the stage of lung development. This typically occurs prior to or after the pseudoglandular stage at 6-16 weeks of gestation 5). In pulmonary hypoplasia, the lung consists of incompletely developed lung parenchyma connected to underdeveloped bronchi. Besides disturbances of the bronchopulmonary vasculature, there is a high incidence, approximately 50-85%, of associated congenital anomalies such as cardiac, gastrointestinal, genitourinary, and skeletal malformations. The diagnosis can result in a spectrum of respiratory complications ranging from transient respiratory distress, chronic respiratory failure, bronchopulmonary dysplasia to neonatal death in very severe cases. Strict diagnostic criteria are not established for pulmonary hypoplasia; various parameters such as lung weight, lung weight to body weight ratio, total lung volume, mean radial alveolar count and lung DNA assessment have been used to classify pulmonary hypoplasia 6).
Your child should be followed by a pediatric pulmonologist after birth so that appropriate diagnostic tests can be performed and routinely followed. Your child’s care and pulmonary hypoplasia treatment plan is based on their specific needs, taking into account each unique diagnosis and treatment your child has received. If early surgery is not performed during infancy, close follow-up of your child is needed. As some cystic lung abnormalities can spontaneously resolve over months to years. Newborns who have been referred for a cystic lesion observed by fetal ultrasonography may have complete resolution on postnatal chest CT. Also, the occurrence of pneumonia or repeated respiratory infections may suggest surgical intervention is needed in a patient who has been conservatively managed.
Various aerosolized medications such as bronchodilators and corticosteroids should be considered if symptoms suggest reactive airway disease or obstructive airway disease.
Persistent pulmonary arterial hypertension can be treated with various pulmonary vasodilators such as inhaled nitric oxide and sildenafil, and endothelin receptor inhibitors such as bosentan.
Pulmonary hypoplasia causes
Pulmonary hypoplasia occurs secondary to a variety of conditions that limit lung development. There are several key factors required for the adequate development of the lung. These are:
- sufficient amniotic fluid volumes
- adequate volume of the thoracic cavity
- normal breathing movement
- normal fluid within the lung
A deficiency in any of these could lead to pulmonary hypoplasia.
For lung development to proceed normally, physical space in the fetal thorax (chest cavity) must be adequate, and amniotic fluid must be brought into the lung by fetal breathing movements, leading to distension of the developing lung. Several studies have demonstrated that gestation age at rupture of membranes (15-28 weeks gestation), latency period (duration between rupture of membranes and birth) and the amniotic fluid index (AFI of less than 1 cm or 5 cm) can influence the development of pulmonary hypoplasia 7).
Most cases of pulmonary hypoplasia are secondary to other congenital anomalies or pregnancy complications. Some cases however can occur as a primary event 8).
With secondary causes, it can result from factors directly or indirectly compromising the thoracic space available for lung growth.
Intrathoracic causes include:
- Congenital diaphragmatic hernia: most common intrathoracic cause
- Congenital cystic adenomatoid malformation (CCAM) or congenital pulmonary airway malformation (CPAM)
- Extralobar sequestration / pulmonary sequestration
- Agenesis of the diaphragm
- Mediastinal mass(es)/tumor(s)
- Mediastinal teratoma
- Thoracic neuroblastomas
- Congenital heart diseases with poor pulmonary (arterial) blood flow
- Tetralogy of Fallot
- Hypoplastic right heart
- Pulmonary artery hypoplasia or unilateral absence of the pulmonary artery
- Scimitar syndrome causing a unilateral right-sided pulmonary hypoplasia
- Trisomies 18 and 21
- Pleural effusions with fetal hydrops, hydrothorax
Extra-thoracic causes include:
- Oligohydramnios and its causes
- Potter sequence: fetal renal anomalies
- Fetal renal agenesis
- Urinary tract obstruction
- Bilateral renal dysplasia
- Bilateral cystic kidneys
- Prolonged rupture of membranes (PROM)
- Preterm premature rupture of membranes (PPROM)
- Skeletal dysplasias, especially those causing a narrow fetal thorax
- Jeune syndrome (asphyxiating thoracic dystrophy)
- Thanatophoric dysplasia
- Achondroplasia
- Achondrogenesis
- Osteogenesis imperfecta
- Short rib polydactyly syndrome
- Campomelic dysplasia
- Large intra-abdominal mass compressing the thorax
- Neuromuscular conditions interfering with fetal breathing movements
- Central nervous system (CNS) lesions
- Lesions of the spinal cord, brain stem, and phrenic nerve
- Neuromuscular diseases (eg, myotonic dystrophy, spinal muscular atrophy)
- Arthrogryposis multiplex congenital secondary to fetal akinesia
- Maternal depressant drugs
Other associations include:
- Fryns syndrome
- Meckel Gruber syndrome
- Neu-Laxova syndrome
- Pena-Shokeir syndrome
Fetal lung fluid and oligohydramnios
Maintenance of fetal lung volume plays a major role in normal lung development. Normal transpulmonary pressure of about 2.5 mm Hg allows the fetal lung to actively secrete fluid into the lumen 9). The effect of stretch of the lung parenchyma induces and promotes lung development. Studies in sheep have demonstrated that tracheal ligation and therefore increased lung distension, accelerates lung growth whereas chronic tracheal fluid drainage has the opposite effect 10). Cohen and colleagues 11) have found that in-utero overexpression of the cystic fibrosis transmembrane conductance regulator (CFTR) increased liquid secretion into the lung, accelerating lung growth in a rat model.
Oligohydramnios is considered to be an independent risk factor for the development of pulmonary hypoplasia. This is likely due to reduced distending forces on the lung. Studies have demonstrated that severe oligohydramnios decreased lung cell size, alters cell shape and may also negatively affect Type 1 cell differentiation which ultimately induces pulmonary hypoplasia.
It has been postulated that the Rho-ROCK pathway can affect the growth of the lung epithelium. Embryonic mouse models have demonstrated that ROCK protein inhibitor decreases the number of terminal lung buds. There are currently several groups studying the role of the Rho/ROCK pathway which has potential therapeutic implications in the reversal of lung hypoplasia 12).
Role of growth factors
Several growth factors such as fibroblast growth factor (FGF), epidermal growth factor (EGF), vascular endothelial growth factor (VEGF) and platelet derived growth factor (PDGF), promote cell proliferation and differentiation. Transforming growth factor family proteins like TGFß1 can oppose these effects.
Embryologically, lungs arise from the foregut. Thyroid transcription factor 1 (TTF-1) is thought to be the earliest embryologic marker associated with cells committed to pulmonary development. FGF signaling is thought to be essential in the formation of TTF-1 expressing cells and this is thought to occur even before the pseudoglandular stage of lung development. Sonic hedgehog (SHH) signaling is further responsible for branching morphogenesis and mesenchymal proliferation. Disruption of any of these pathway may result in primary pulmonary hypoplasia 13).
FGF7 and FGF10 promote epithelial proliferation and formation of the bronchial tree. Overexpression of FGF10 can also stimulate the formation of cysts in the rat lung 14). EGF promotes lung branching and Type II alveolar cell proliferation. PDGF plays a crucial role in alveolarization. VEGF promotes angiogenesis and the differentiation of embryonic mesenchymal cells into endothelial cells. Bone morphogenetic protein was thought to oppose lung growth; however recent data suggests that in the presence of mesenchymal cells, BMP4 is a potent inducer of tracheal branching 15). Aberrant expression of these growth factor proteins in the amniotic fluid during pregnancy have been implicated in abnormal lung development. Interestingly, higher concentrations of VEGF are seen in the amniotic fluid in the second and third trimester and may be a molecular marker for hypoxia which requires further investigation 16).
Congenital diaphragmatic hernia
The pathogenesis of pulmonary hypoplasia associated with congenital diaphragmatic hernia remains unclear. Several mechanisms have been suggested. The nitrofen model of congenital diaphragmatic hernia is widely accepted. Nitrofen is a human carcinogen and the retinoid acid signaling pathway is essential for the normal development of the diaphragm. Perturbation of this pathway with compounds such as nitrofen, can induce congenital diaphragmatic hernia and pulmonary hypoplasia. Esumi and colleagues demonstrated that that administration of insulin-like growth factor 2 (IGF2) to nitrofen-induced hypoplastic lungs lead to alveolar maturation 17). Furthermore, recent data suggests that prenatal treatment with retinoic acid results in increased levels of placental IGF2 and promotes both placental and fetal lung growth in nitrofen induced congenital diaphragmatic hernia 18).
Interestingly, erythropoietin (EPO) is a direct target of retinoic acid. A recent study has demonstrated decreased levels of erythropoietin mRNA in the liver and kidney of rats which may explain modifications in the pulmonary vasculature in congenital diaphragmatic hernia 19).
A recent study has also suggested a possible role of interleukin 6 (IL-6) in inducing catch-up growth particularly in nitrofen pre-treated explant fetal rat lungs 20).
In cases of congenital diaphragmatic hernia (congenital diaphragmatic hernia) associated with pulmonary hypoplasia, hypertrophy of the contralateral lung has been demonstrated, with associated pulmonary artery hypertension. The hypoxemia in pulmonary hypoplasia stems from hypoventilation and right-to-left extrapulmonary shunting.
Pulmonary hypoplasia symptoms
There is wide variation in the clinical presentation of pulmonary hypoplasia, depending on the extent of hypoplasia and other associated anomalies 21).
The history may include poor fetal movement or amniotic fluid leakage and oligohydramnios. The neonate may be asymptomatic or may present with severe respiratory distress or apnea that requires extensive ventilatory support. In older children, dyspnea and cyanosis may be present upon exertion, or a history of repeated respiratory infections may be noted.
The external chest may appear normal or may be small and bell shaped, with or without scoliosis. A mediastinal shift is observed toward the involved side, and dullness upon percussion is heard over the displaced heart. In right-sided hypoplasia, the heart is displaced to the right, which may lead to a mistaken diagnosis of dextrocardia. Breath sounds may be decreased or absent on the side of hypoplasia, especially over the bases and axilla.
Some infants may present with otherwise asymptomatic tachypnea, and other may have severe respiratory distress at birth requiring ventilatory support. Pneumothorax, either spontaneous or associated with mechanical ventilation, may occur.
Infants with secondary pulmonary hypoplasia may have associated congenital anomalies or features suggestive of neuromuscular diseases. Such patients may have myopathic facies, with a V-shaped mouth, muscle weakness, and growth restriction. Multiple genetic syndromes associated with primary pulmonary hypoplasia are reported in the literature such as Scimitar Syndrome, Trisomy 21, and Pena-Shokeir Syndrome (fetal akinesia) 22).
Compression deformities due to prolonged oligohydramnios, contractures, and arthrogryposis may be present. The Potter facies (hypertelorism, epicanthus, retrognathia, depressed nasal bridge, low set ears) suggest the possibility of lung hypoplasia caused by the associated renal defects.
Abdominal masses, such as cystic renal diseases and an enlarged bladder, must be sought. Associated anomalies of the cardiovascular, gastrointestinal (eg, tracheoesophageal fistula, imperforate anus, communicating bronchopulmonary foregut malformation), and genitourinary systems, as well as skeletal anomalies of the vertebrae, thoracic cage, and upper limbs, may be found upon examination 23).
Pulmonary hypoplasia complications
Complications in pediatric pulmonary hypoplasia are as follows:
- Mortality due to acute respiratory failure in the neonatal period
- Chronic respiratory failure or insufficiency
- Pneumothorax, either spontaneous or as a result of ventilatory support
- Persistent pulmonary hypertension caused by a reduced pulmonary vascular bed and worsened by hypoxia or a coexisting left-to-right intracardiac shunt
- Chronic lung disease of infancy caused by prolonged ventilatory support
- Airway abnormalities, including tracheobronchial compression and tracheomalacia caused by the displaced aorta and enlarged left pulmonary artery
- Restrictive lung disease due to reduced total lung capacity
- Recurrent respiratory infections
- Recurrent wheezing episodes
- Reduced exercise tolerance
- Scoliosis in adolescent years due to abnormal thoracic cage development
- Nutritional, musculoskeletal, neurological, and gastrointestinal comorbidities
- Delayed growth and development
Pulmonary hypoplasia diagnosis
If the cause of the pulmonary hypoplasia is renal pathology, serum creatinine, blood urea, and electrolytes levels should be measured to assess renal function.
Radiographs
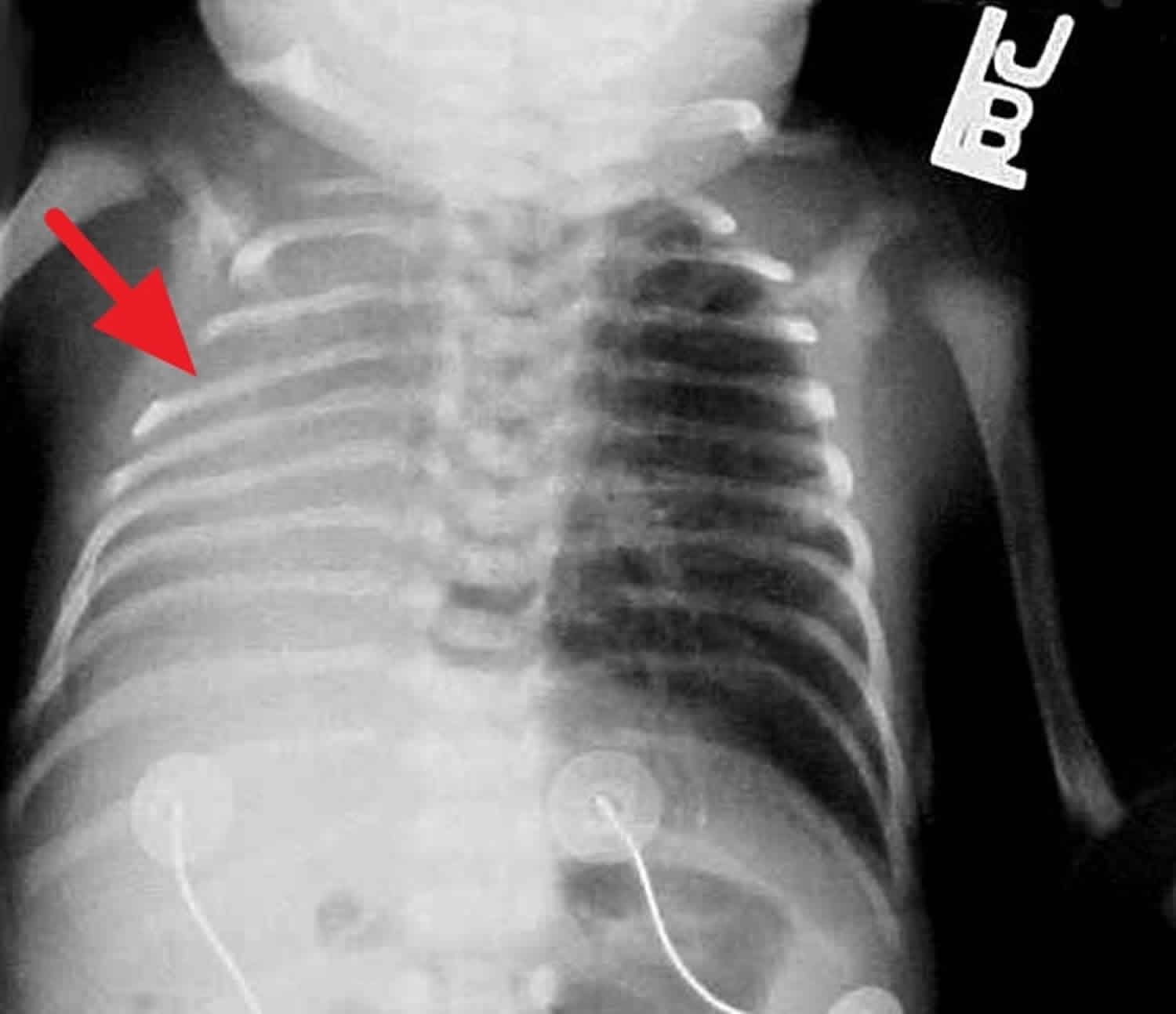
Chest radiographic findings vary. The ribs may appear crowded with a low thoracic-to-abdominal ratio, and the chest wall is classically bell-shaped; however, lung fields are clear unless there is also coexisting respiratory distress syndrome. Pneumothorax or other forms of air leak are frequently present. Films may also show features of the neonate’s underlying condition. In severe cases, there may be mediastinal shift with a homogenous density on the involved hypoplastic side and compensatory herniation of the contralateral lung across the mediastinum. Rib deformities may be observed. See the images below.
2D and 3D ultrasonography
Thoracic circumference (TC), thoracic circumference to abdominal circumference (TC:AC) ratio and lung area (LA) are frequently used measurements to assess prenatal risk for pulmonary hypoplasia 24). Thoracic circumference (TC) and lung area (LA) are gestational-age dependent whereas TC:AC ratio is not affected by gestational age. All of these measurements have a high specificity (between 40-100%) but none have a high enough sensitivity to be used reliably in clinical practice. However, in fetuses with congenital diaphragmatic hernia, the observed to expected lung-to-head ratio (LHR) measured by two-dimensional ultrasound remains the best predictor of pulmonary hypoplasia 25).
Three-dimensional ultrasound techniques, which include pulmonary volume measurement, appears to have a high sensitivity 92% and specific 84% and appears to be a reliable technique to predict pulmonary hypoplasia 26). Barros and Colleagues 27) found that lung volumes measurements using 3D ultrasound has a high sensitivity (83.3%) and specificity (100%) for predicting lethal pulmonary hypoplasia in infant with skeletal anomalies.
Targeted fetal ultrasonography may demonstrate renal malformations, oligohydramnios, and decreased fetal movements in fetal neuromuscular diseases. While this is readily available at most centers, diagnosing disease requires expertise and can be limited by the presence of maternal obesity, low amniotic fluid index (AFI) and fetal malposition 28).
Autopsy studies have shown that pulmonary hypoplasia is associated with a reduction in a number of pulmonary vessels and with increased arterial smooth muscle thickness, which may lead to increased pulmonary vascular resistance and decreased pulmonary arterial compliance. Pulmonary vasculature remodeling and pulmonary hypertension is particularly common in congenital diaphragmatic hernia and results in high mortality 29). Given abnormalities in the vasculature, Doppler ultrasonography has been studied as way to predict pulmonary hypoplasia. Determination of pulmonary artery blood velocity waveforms is one tool used to diagnose pulmonary hypoplasia, however as a single test is unreliable. Pulsatile index of the ductus arteriosus for predicting pulmonary hypoplasia had a sensitivity of only 37% and specificity of 2% which is again, not clinically reliable 30).
Other imaging studies
Echocardiography may be used to identify associated cardiac anomalies. The frequency of cardiovascular malformations associated with isolated congenital diaphragmatic hernia is 11-15% 31). The most common anomalies include atrial and ventricular septal defects, conotruncal defects, and left ventricular outflow tract obstructive defects.
Angiography is indicated to confirm the diagnosis of any aberrant pulmonary vessels, to rule out scimitar syndrome, and to confirm reduced pulmonary vascular bed.
MRI or magnetic resonance angiography (MRA) may also be used to identify the smaller pulmonary arterial supply to the affected lung and the presence of other abnormal vascular anatomy.
Both MRI and ultrasonography appear to be useful in determining the degree of pulmonary hypoplasia 32). Particularly in congenital diaphragmatic hernia, MRI based total fetal lung volume (FLV) and fetal body volume (FBV) measurements are useful in predicting post-natal survival. Recent studies are also demonstrating that MRI based FLV:FBV ratio measurements are not only able to predict neonatal mortality but also able to predict extracorporeal membrane oxygenation (ECMO) requirement with high accuracy 33).
Lung scintigraphy has been used to evaluate the degree of pulmonary hypoplasia in infants with congenital diaphragmatic hernia. One study suggested that lung scintigraphy is useful to predict long-term pulmonary morbidity and poor nutritional status in survivors of congenital diaphragmatic hernia 34).
Other tests
Obtaining an ECG and/or echocardiogram is important to distinguish between dextrocardia and dextroposition caused by pulmonary hypoplasia. In dextrocardia, ECG findings include an inverted P wave and T in lead 1, with negative QRS deflection and a reverse pattern between aVR and aVL. A mirror image progression is observed from V1 to a right-sided V6 lead. A tall R in lead V1 or an RS ratio equal to or greater than 1 also suggests dextrocardia.
The frequency of cardiovascular malformations associated with isolated congenital diaphragmatic hernia is 11-15% 35). The most anomalies include atrial and ventricular septal defects, conotruncal defects, and left ventricular outflow tract obstructive defects.
Bronchoscopy or bronchography is indicated because the reduced size of a bronchus and its branches confirms the diagnosis.
Pulmonary function testing is difficult to obtain in the young age, however it may a useful tool in monitoring the course of the disease to assess lung maturation and development. A recent study has demonstrated that lung function remains abnormal in the first three years of life in children with congenital diaphragmatic hernia. This study revealed normalization of total lung capacity, however with increasing residual volumes likely due to pulmonary overinflation. They hypothesized that the pulmonary hyperinflation was not due to normal alveolarization that occurs in the first three years of life, but is likely due to overdistended, simplified air spaces that was functionally different from those seen in normally grown lungs 36). Another study reported normalization of all lung function parameters after surgery by age 24 months 37). As expected, lung function significantly correlated with increase in age, height, and, especially, weight.
Histologic findings
On autopsy, in pulmonary hypoplasia, the overall lung size is reduced, cell numbers are decreased, branches of airways may be narrower and fewer, alveolar differentiation may be reduced, and a surfactant deficiency may be present.
Histopathologic descriptions of pulmonary hypoplasia may have limited value since some may appear similar to normal lung. However, in other cases there may be a reduction in a number of pulmonary vessels (and smaller pulmonary arterioles) and increased arterial smooth muscle thickness, indicating the presence of pulmonary hypertension.
The diagnosis of pulmonary hypoplasia is made if the lung weight–to–body weight ratio is less than 0.015 in infants born before 28 weeks of gestation and less than 0.012 in infants born after 28 weeks of gestation, in conjunction with a mean radial alveolar count (RAC) of less than 4%. The radial alveolar count (RAC) provides a simple objective measurement of the “relative paucity of alveoli” or “crowding of bronchial structures, which is unaffected by the state of expansion of the lungs.
In addition, in infants with severe risk factors (renal anomalies, diaphragmatic hernia), lung hypoplasia may be diagnosed by a lung volume–to–body weight ratio less than the 10th percentile when assessed on age-specific reference values 38).
Pulmonary hypoplasia treatment
In fetuses with pulmonary hypoplasia, interventions can be done prenatally and treatment goals should be established for postnatal care. Prenatal interventions are performed with the goal of delaying preterm labor and allowing for lungs to mature.
Preterm rupture of membranes without signs of fetal distress or intrauterine infection is treated conservatively with or without tocolytics, antibiotics, and steroids in various combinations. Antenatal corticosteroids enhance fetal lung maturation in pregnancies less than 34 weeks of gestation. If gestational age is uncertain, lung maturity can be determined by aspiration of amniotic fluid from the vaginal vault. The lamellar body counts are a direct measurement of surfactant production by type II pneumocytes. If this initial screen shows neither clearly mature nor immature fetal lung, then the lecithin/sphingomyelin (L/S) ratio can be determined from amniotic fluid. The risk of respiratory distress is very low when the L/S ratio is greater than 2.0.
Amnioinfusions and amniopatch techniques have shown promising results in the treatments of preterm labor. Amnioinfusion consists of instilling isotonic fluid into the amniotic cavity. Amniopatch consists of intraamniotic injection of platelets and cryoprecipitate with the goal of sealing amniotic fluid leak. Small cases series have reported that both techniques reduce perinatal complications and prolong pregnancy particularly in severe oligohydramnios 39).
After delivery, the infant needs respiratory support, which can range from supplying supplemental oxygen to mechanical ventilation, including high-frequency ventilation and extracorporeal membrane oxygenation (ECMO). Ventilatory strategies that have veered toward the use of gentle volume recruitment, permissive hypercapnia, especially in cases of congenital diaphragmatic hernia (congenital diaphragmatic hernia), have led to increased survival and improved outcomes 40). Fetal MRI based lung volume assessment may be useful in predicting the severity of pulmonary hypoplasia and may also predict the need for ECMO. Weidner and colleagues demonstrated lower FLV:FBV ratios in infants who required ECMO 41). While the timing of congenital diaphragmatic hernia repair for infant on ECMO remains controversial, there are studies that show that surgical repair of congenital diaphragmatic hernia while on ECMO, can be done safely and is associated with good survival and there may be increased mortality associated with delayed repair 42). Pulmonary hypertension contributes to significant mortality in patient with congenital diaphragmatic hernia and this particular subset of patients may have additional benefit from early ECMO support.
There is conflicting data regarding the efficacy of inhaled nitric oxide (iNO) to manage pulmonary hypertension secondary to congenital diaphragmatic hernia. Randomized controlled trials of inhaled nitric oxide (iNO) treatment for infants with congenital diaphragmatic hernia have shown marginal, if any, efficacy. Poor left ventricular function and/or left ventricular hypoplasia may account for some of the poor response to iNO. Infants with severe respiratory failure secondary to pulmonary hypoplasia and documented persistent pulmonary hypertension of the newborn may benefit from iNO, but the data are limited 43). Aggressive ventilation in these infants causes overexpansion of lungs with compresses intra-alveolar capillaries which further aggravates pulmonary hypertension. If this is the case, hemodynamics should be optimized prior to initiating nitric oxide. More recently there are smaller population studies that show that nitric oxide may be beneficial as adjunct therapy in combination with Sildenafil and dopamine infusions to improve survival, but larger studies are needed 44).
Low lung compliance associated with congenital diaphragmatic hernia is thought to be secondary to surfactant deficiency, although there is very limited and conflicting data regarding this theory. One study shows that infants with congenital diaphragmatic hernia had lower rates of synthesis of surfactant protein B (SP-B) and less SP-B in tracheal aspirates compared with age-matched controls without lung disease 45). While surfactant is not contraindicated, it does not seem to provide additional survival benefit in infants with congenital diaphragmatic hernia.
Of note, overexpansion of hypoplastic lungs compresses intra-alveolar capillaries and aggravates pulmonary hypertension. Partial liquid ventilation has also been used; however, data are lacking to support or refute the use of partial liquid ventilation in children with acute lung injury or acute respiratory distress syndrome 46).
Dialysis for support of renal function is provided in some cases, but it should be started only after careful consideration. Patients with severe chronic renal impairment with pulmonary hypoplasia have a poor prognosis; the ultimate outcome is difficult to improve, even with optimal renal and respiratory support.
Some studies suggest that strict infection control may improve the outcome of neonates with congenital diaphragmatic hernia without the need for extracorporeal membrane oxygenation (ECMO) 47).
Medical management of cystic adenomatoid malformations (CCAM) and prognosis is dependent on the size of the lesion. Microcysts 58</ref> Spontaneous improvement and possible resolution may occur over months to years in many of these lesions 48). Their management must be individualized, with very large lesions resulting in lung hypoplasia or fetal hydrops required possible fetal surgery 49). In most cases of fetal lung lesions, continued observation with possible postnatal therapy occurs if respiratory distress or failure to thrive develops 50).
Multiple studies have proven the importance of the retinoic acid signaling pathway in lung development as mentioned above. In keeping with this, the role of retinoic acid supplementation and antioxidants in pulmonary hypoplasia has been extensively studied. There is some promising human data that demonstrated decrease in incidence of bronchopulmonary dysplasia in extremely low birth infants who received vitamin A supplementation 51). There are also several animal models that show an increase in VEGF expression and increased lung alveolarization in response to vitamin A supplementation. Despite encouraging in vitro work, supplementation with vitamin A failed to reverse oligohydramnios-induced pulmonary hypoplasia in fetal rats.
Surgical treatment
A multidisciplinary team with expertise in fetal surgery should be involved, when feasible, in all cases of severe pulmonary hypoplasia. A major indication for fetal surgery is the presence of hydrops and a gestation of less than 32 weeks. In general cases that require surgical intervention are large cystic lung malformations and congenital diaphragmatic hernias.
Cystic lung malformations
Thoracocentesis or thoracoamniotic shunts can allow for drainage of fluid from the congenital cystic adenomatoid malformation (CCAM), but the fluid usually rapidly re-accumulates. Thoracoamniotic shunts may be offered in pregnancies complicated by hydrops secondary to the presence of a large or multiple communicating macrocysts or severe pleural effusions. Shunt placement has been reported to decrease congenital cystic adenomatoid malformation (CCAM) mass volumes by an average of 50%, and as much as 80% in some cases 52).
In cases of significant mass effect due to congenital congenital cystic adenomatoid malformations (CCAMs) (or other solid lung mases) recommendations for delivery can range from an ex utero intrapartum treatment (EXIT) procedure with tumor resection while still on placental bypass, to elective cesarean delivery and immediate pediatric surgical evaluation and resection, to delivery with on-site pediatric surgical services. In cases in which masses plateau earlier in their growth phase, and presents a nonsignificant risk of pulmonary hypoplasia or hemodynamic compromise, surgery can be planned as an outpatient at age 4-6 week 53). Therefore, the management of the congenital cystic lung abnormalities needs to consider the spontaneous improvement and possible resolution that occurs over months to years in many of these lesions 54). Up to 15% of prenatally diagnosed congenital cystic adenomatoid malformations (CCAMs) regress and may sonographically “disappear” by becoming isoechoic within the surrounding normal lung tissue. However, these lesions can still be identified on postnatal CT scan with contrast.
The risks of subsequent malignant degeneration of congenital cystic adenomatoid malformations (CCAMs) are poorly understood. After removal by lobectomy, the remaining normal ipsilateral lung demonstrates compensatory lung growth, and in general these children have no residual respiratory problems 55).
Thoracocentesis or thoracoamniotic shunts can allow for drainage of fluid from the congenital cystic adenomatoid malformation (CCAM), but the fluid usually rapidly reaccumulates. Thoracoamniotic shunts may be offered in pregnancies complicated by hydrops secondary to the presence of a large or multiple communicating macrocysts or severe pleural effusions. Shunt placement has been reported to decrease congenital cystic adenomatoid malformation (CCAM) mass volumes by an average of 50%, and as much as 80% in some cases 56).
Intrauterine vesicoamniotic shunts and endoscopic ablation of posterior urethral valves are other techniques that are currently used in fetuses with urinary tract obstruction and pulmonary hypoplasia. With careful case selection, pulmonary hypoplasia is prevented, and postnatal renal and respiratory function is improved 57).
Congenital diaphragmatic hernia
In experimental animals, percutaneous fetal endoluminal tracheal occlusion (FETO) induces lung growth and morphologic maturation. Fetal endoluminal tracheal occlusion (FETO) with a clip may lead to accelerated lung growth and prevent pulmonary hypoplasia. Fetal endoluminal tracheal occlusion (FETO) is currently being studied at some centers across Europe, as a way to improve survival in cases of pulmonary hypoplasia associated with severe congenital diaphragmatic hernia 58). There are variations in the technique but most centers prefer the non-invasive technique, where a balloon, inserted into the tracheal lumen at 22-28 weeks’ gestation 59). Balloon occlusion creates a transpulmonic pressures, prevent fluid egress of fluid from the fetal lung which stimulate lung growth. It has been suggested that later insertion of the balloon beyond 29 weeks does not results in significant lung growth 60). A recent study from Texas has reported improved postnatal outcomes in infants with severe congenital diaphragmatic hernia 61). This procedure was found to be minimally invasive, may reverse pulmonary hypoplasia changes, and may improve survival rate in these highly selected cases. In addition, the airways can be restored before birth.
The optimal time of surgery for congenital diaphragmatic hernia repair varies from center to center. Surgical repair typically involves primary or patch closure of the diaphragm through an open abdominal approach. Successes have been reported with an endoscopic approach; however, it is associated with an increased incidence of hernia recurrence 62). The decision is made based on the severity of the lesion, hemodynamics of the patient and the center’s preferences. Intraoperative considerations include the length of operative time, as thoracoscopic repair is associated with substantially longer operative times, leading to concerns for intraoperative instability, carbon dioxide retention, and pulmonary vasospasm for patients with moderate-to-severe pulmonary hypertension 63). There are some studies that suggest that early intervention in patients on ECMO, reduced the total duration of ECMO, reduced surgical complications and increased survival. However, this data may be skewed toward patients who may have been too sick to be weaned off ECMO prior to surgery and further studies are needed 64).
Follow up care
Since chronic lung disease is common in survivors of pulmonary hypoplasia, these infants and children have an increased risk of fatality and serious morbidity from upper respiratory tract infections (URTIs) and lower respiratory tract infections (LRTIs). Antiviral and antibiotics should be administered based on clinical symptoms and signs.
Children may be given bronchodilators and/or inhaled corticosteroids for the treatment of wheezing episodes and/or reactive airway disease.
Respiratory syncytial virus (RSV) prophylaxis should be considered during RSV season in infants younger than two years who have been treated with oxygen or medication for chronic lung disease within 6 months of the start of RSV season. Palivizumab is a humanized monoclonal antibody (IgG) directed against the fusion protein of RSV and has been shown to reduce the risk of hospitalization from RSV infection in high-risk pediatric patients by 55%. RSV season in most parts of the United States is from October to March. The dose is 15 mg/kg via intramuscular injection monthly throughout RSV season.
Children with pulmonary hypoplasia should receive the influenza vaccine at the start of every influenza season, which in the United States, while varying from season to season, begins as early as October. The influenza season peaks in January or February and continues as late as May.
Children with chronic lung disease are considered at high risk for invasive pneumococcal disease. If younger than two years, they should be administered the 13-valent pneumococcal conjugate vaccine (PCV13) 4-dose series at ages two, four, and six months, with a booster dose at 12-15 months. If aged 24 months to five years, they should receive 1 or 2 doses of PCV13 if they have not already completed the 4-dose series. Anyone over the age of two, with chronic lung disease, should also receive 1 dose of PCV23.
Pulmonary hypoplasia prognosis
Mortality has traditionally been very high. In a retrospective study of 76 premature infants less than 35 weeks’ gestation, 20 had prolonged rupture of membrane of more than 5 days and were clinically diagnosed with pulmonary hypoplasia. Of those 20 infants with pulmonary hypoplasia, 18 died. In another retrospective study of 117 infants of less than 37 weeks’ gestation who had prolonged rupture of membrane of more than 99 hours, 11 died and were considered to have pulmonary hypoplasia. The median age of death was 20 hours (range, 12-48 hours), mostly commonly from respiratory failure.
In different studies, mortality rates associated with pulmonary hypoplasia are reported to be as high as 71-95% in the perinatal period 65).
The following conditions increase the risk of mortality 66):
- Earlier gestational age at rupture of membranes, particularly at less than 25 weeks of gestation
- Severe oligohydramnios (amniotic fluid index < 4) for more than 2 weeks
- Earlier delivery (decreased latency period)
- Right-sided lesion
- Presence of genetic anomalies
To avoid mortality from severe lung hypoplasia in association with congenital diaphragmatic hernia or congenital cystic adenomatoid malformation (CCAM), fetal surgical intervention has been attempted. Most studies report a mortality rate of 25-30% in neonates with congenital diaphragmatic hernia andcongenital cystic adenomatoid malformation (CCAM) at high volume centers; mortality can be as high as 45% at peripheral care centers. However, in other cystic lung lesions, most are clinically asymptomatic and may not need aggressive management 67).
Risk factors for a poor outcome include the presence of hydrops fetalis, with a mortality rate as high as 80-90%. Other indicators include the type of congenital cystic adenomatoid malformation (CCAM) and its size. All of these factors reflect the degree of pulmonary compromise with lesions that result in varying degrees of pulmonary hypoplasia.
There is a recent retrospective study from Barcelona that studied 60 cases of pulmonary hypoplasia between 1995 to 2014, that found a mortality rate of 47% in the first 60 days of life and up to 75% in the first day of life 68).
While antepartum amnioinfusions for treatment of oligohydramnios have significantly reduced the risk of pulmonary hypoplasia, longitudinal follow-up studies are lacking on the long-term outcomes of these children.
Of children with pulmonary hypoplasia secondary to congenital diaphragmatic hernia, the postnatal survival rate of congenital diaphragmatic hernia at tertiary centers has improved, with reported rates of 70-92% 69). However, the survival rates do not account for the cases of congenital diaphragmatic hernia that are stillborn, died outside a tertiary center, or died as a result of spontaneous or therapeutic abortion.
Congenital diaphragmatic hernia survivors have a high incidence of respiratory, nutritional, musculoskeletal, neurological, and gastrointestinal morbidities 70). In a prospective study of 41 congenital diaphragmatic hernia survivors, abnormal muscle tone was found in 90% at age 6 months and 51% at age 24 months. While almost half (49%) had normal scores for neurocognitive and language skills, 17% had mildly delayed and 15% had severe delayed scores. Likewise, in psychomotor testing, while 46% had normal scores, 23% and 31% scored as mildly delayed and severely delayed, respectively. Autism was present in 7%. Studies of brain maturation using MRI show delayed structural brain development and other abnormalities that may lead to long-term neurologic complications 71).
In a retrospective follow-up study of 55 children survivors with scimitar syndrome followed at one center, a high rate of respiratory complications was observed. All (100%) of the children had right lung hypoplasia of varying degrees of severity. The median duration of follow-up was 7.2 years. Pulmonary infections were reported in 38%, and 43% of children reported wheezing episodes during the last 12 months of follow-up. A restrictive pattern of lung function was observed in the majority of patients, likely related to right-sided lung hypoplasia. Lower total lung capacity values were seen in children with the infantile form of scimitar syndrome, possibly reflective of the severity of pulmonary hypoplasia in these children 72).
Right-sided hypoplasia, typically secondary to right sided congenital diaphragmatic hernia, seems to carry a higher mortality. This is likely due to higher risk of recurrent herniation, increased risk of pulmonary complications, requiring pulmonary vasodilator therapy and tracheostomy. However, no differences in neurodevelopmental outcomes was found 73).
A minimum lung volume of 45% compared with age-matched control subjects has been shown to be a predictor of survival in neonates with diaphragmatic hernia treated with extracorporeal membrane oxygenation (ECMO). Similarly, a functional residual capacity of 12.3 mL/kg, about one half the normal capacity, has been thought to be a predictor of survival in pulmonary hypoplasia with congenital diaphragmatic hernia.
References [ + ]


{kind=link}