Contents
Diastrophic dysplasia
Diastrophic dysplasia also called diastrophic dwarfism, is a type of short limb skeletal dysplasia (micromelic dwarfism) that is present at birth (congenital). The word “dysplasia” refers to abnormal growth. Diastrophic dysplasia is a disorder of cartilage and bone development that leads to an onset of joint pain and deformity. Diastrophic dysplasia is a rare genetic condition that causes short stature and unusually short arms and legs (short-limbed dwarfism), where a child’s legs and arms do not grow and develop to the typical adult length. Adult patients have a stature between 100 and 140 cm. Other characteristics of diastrophic dysplasia are abnormal development of bones (skeletal dysplasia) and joints (joint dysplasia) in many areas of the body; progressive abnormal curvature of the spine (scoliosis and/or kyphosis); abnormal tissue changes of the outer, visible portions of the ears (pinnae); and/or, in some cases, malformations of the head and facial (craniofacial) area. Typically there is limb shortening, hitchhiker thumbs, spinal deformity, and large joint contractures with associated deformities and premature degenerative disease. Other classic findings include ulnar deviation of the fingers, a large sandal gap (space between first and second pedal digits), and clubfoot. However, the range and severity of associated symptoms and physical findings may vary greatly from case to case.
Diastrophic dysplasia is characterized by limb shortening, normal-sized skull, hitchhiker thumbs, spinal deformities (scoliosis, exaggerated lumbar lordosis, cervical kyphosis), and contractures of the large joints with deformities and early-onset osteoarthritis. Joint contractures and spinal deformity tend to worsen with age 1). Other typical findings are ulnar deviation of the fingers, gap between the first and second toes, and clubfoot 2). On occasion diastrophic dysplasia can be lethal at birth, but most affected individuals survive the neonatal period and develop physical limitations with normal intelligence 3). Occasionally, children with diastrophic dysplasia die in infancy due to respiratory complications such as pneumonia.
At birth, babies with diastrophic dysplasia tend to be shorter, usually about 16.5 inches long. On average, most newborns are between 19 and 21 inches long.
In most infants with diastrophic dysplasia, the first bone within the body of each hand (first metacarpals) may be unusually small and “oval shaped,” causing the thumbs to deviate away (abduction) from the body (“hitchhiker thumbs”). Other fingers may also be abnormally short (brachydactyly) and joints between certain bones of the fingers (proximal interphalangeal joints) may become fused (symphalangism), causing limited flexion and restricted movement of the finger joints. Affected infants also typically have severe foot deformities (talipes or “clubfeet”) due to abnormal deviation and fusion of certain bones within the body of each foot (metatarsals). In addition, many children with the disorder experience limited extension, partial (subluxation) or complete dislocation, and/or permanent flexion and immobilization (contractures) of certain joints.
In most infants with diastrophic dysplasia, there is also incomplete closure of bones of the spinal column (spina bifida occulta) within the neck area and the upper portion of the back (lower cervical and upper thoracic vertebrae). In addition, during the first year of life, some affected children may begin to develop progressive abnormal sideways curvature of the spine (scoliosis). During adolescence, individuals with the disorder may also develop abnormal front-to-back curvature of the spine (kyphosis), particularly affecting vertebrae within the neck area (cervical vertebrae). In severe cases, progressive kyphosis may lead to difficulties breathing (respiratory distress). Some individuals may also be prone to experiencing partial dislocation (subluxation) of joints between the central areas (bodies) of cervical vertebrae, potentially resulting in spinal cord injury. Such injury may cause muscle weakness (paresis) or paralysis and/or life-threatening complications.
In addition, most newborns with diastrophic dysplasia have or develop abnormal fluid-filled sacs (cysts) within the outer, visible portions of the ears (pinnae). Within the first weeks of life, the pinnae become swollen and inflamed and unusually firm, thick, and abnormal in shape. Over time, the abnormal areas of tissue (lesions) may accumulate deposits of calcium salts (calcification) and eventually develop into bone (ossification). Some affected infants may also have abnormalities of the head and facial (craniofacial) area including incomplete closure of the roof of the mouth (cleft palate) and/or abnormal smallness of the jaws (micrognathia). In addition, in some affected infants, abnormalities of supportive connective tissue (cartilage) within the windpipe (trachea), voice box (larynx), and certain air passages in the lungs (bronchi) may result in collapse of these airways, causing life-threatening complications such as respiratory obstruction and difficulties breathing. In some individuals with the disorder, additional symptoms and physical findings may also be present.
Children with diastrophic dysplasia have characteristic craniofacial features (Figure 1).
Children born with diastrophic dysplasia may do physical things — like rolling over, sitting up and walking — later than other children.
Diastrophic dysplasia is caused by mutations in the SLC26A2 gene and is inherited in an autosomal recessive manner 4). Diastrophic dysplasia occurs equally in males and females and most often in whites. Although the exact prevalence of diastrophic dysplasia is unknown, researchers estimate that it affects about 1 in 500,000 newborns in the United States. Diastrophic dysplasia is more common in Finland, where it affects about 1 in 33,000 newborns 5).
Management of diastrophic dysplasia consists of maintaining joint position and mobility through physical therapy and casting. Surgical correction of clubfoot may be necessary. Arthroplasty of the hips and knees to decrease pain and increase motility may also be indicated. Indications for surgical correction of scoliosis have not yet been established 6).
Figure 1. Diastrophic dysplasia
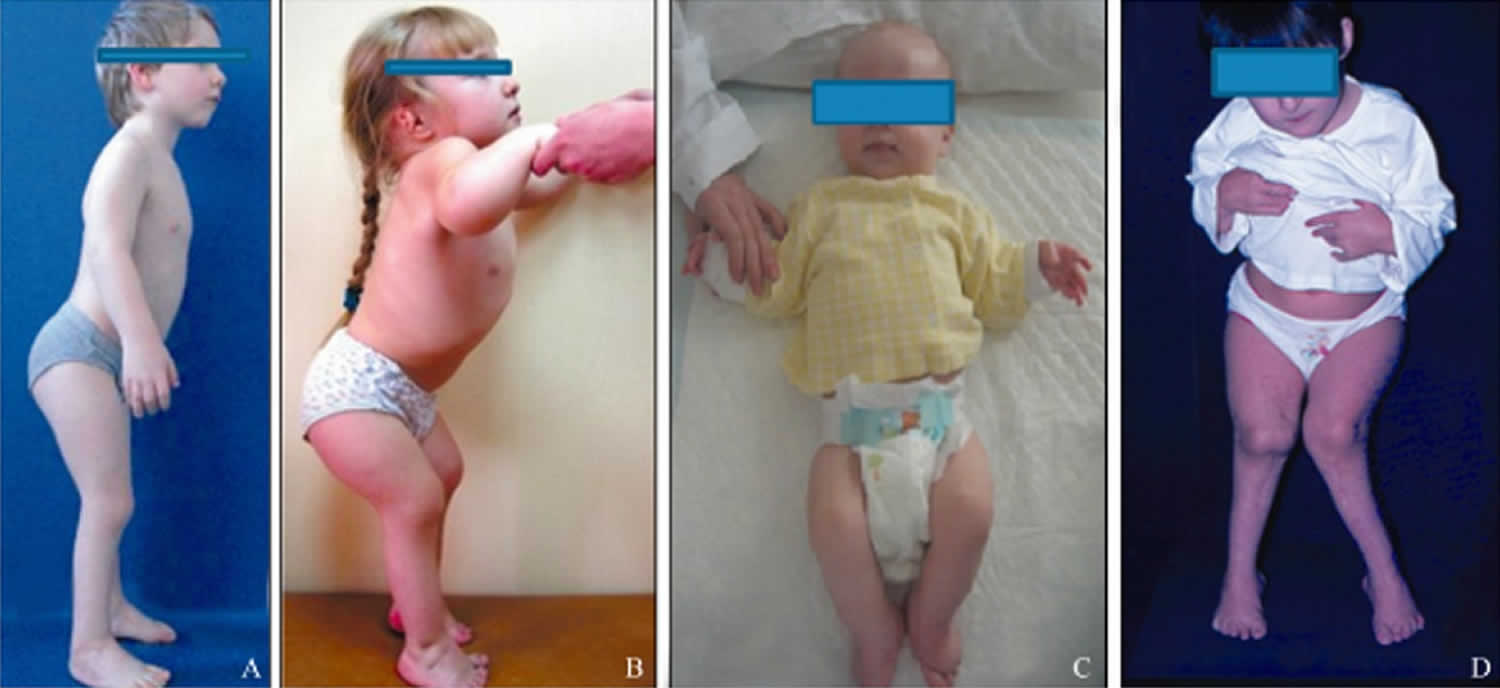
Footnote: The clinical appearances of children with diastrophic dysplasia showing the characteristic craniofacial features (A–C). Figure 1A shows a 10‐year‐old boy with severe short stature, thickened ear lobes and multiple contractures (elbows, pelvis, and knees). Figure 1B shows a 2‐year‐old girl with severe short stature, thickened ear lobes and swellings of the pinnae associated with narrow external auditory canals. Figure 1C shows a 6‐month‐old boy with micromelic dwarfism, a characteristic craniofacial contour and contractures of the elbows and knees. Figure 1D shows a 12‐year‐old girl whose knees had been operated on to relieve the contractures; however, the contractures had recurred.
[Source 7) ]Diastrophic dysplasia causes
Diastrophic dysplasia is one of several skeletal disorders caused by mutations in the SLC26A2 gene also called DTDST gene (diastrophic dysplasia sulfate transporter gene) that has been located on the long arm (q) of chromosome 5 (5q32-q33.1). The SLC26A2 gene or DTDST gene provides instructions for making a protein that transports charged molecules (ions), particularly sulfate ions, across cell membranes. This protein appears to be active in many of the body’s tissues, including developing cartilage. Cartilage is a tough, flexible tissue that makes up much of the skeleton during early development. Most cartilage is later converted to bone, except for the cartilage that continues to cover and protect the ends of bones and is present in the nose and external ears.
Cartilage cells use sulfate ions transported by the SLC26A2 protein to build molecules called proteoglycans. These molecules, which each consist of several sugars attached to a protein, help give cartilage its rubbery, gel-like structure. Because sulfate ions are required to make proteoglycans, the transport activity of the SLC26A2 protein is essential for normal cartilage formation.
Mutations in the SLC26A2 gene or DTDST gene impede cellular incorporation of sulfate and result in production of undersulfated cartilage proteoglycans, disrupting the assembly of cartilage matrix, preventing bones from forming properly and resulting in the skeletal problems characteristic of diastrophic dysplasia 8).
Diastrophic dysplasia inheritance pattern
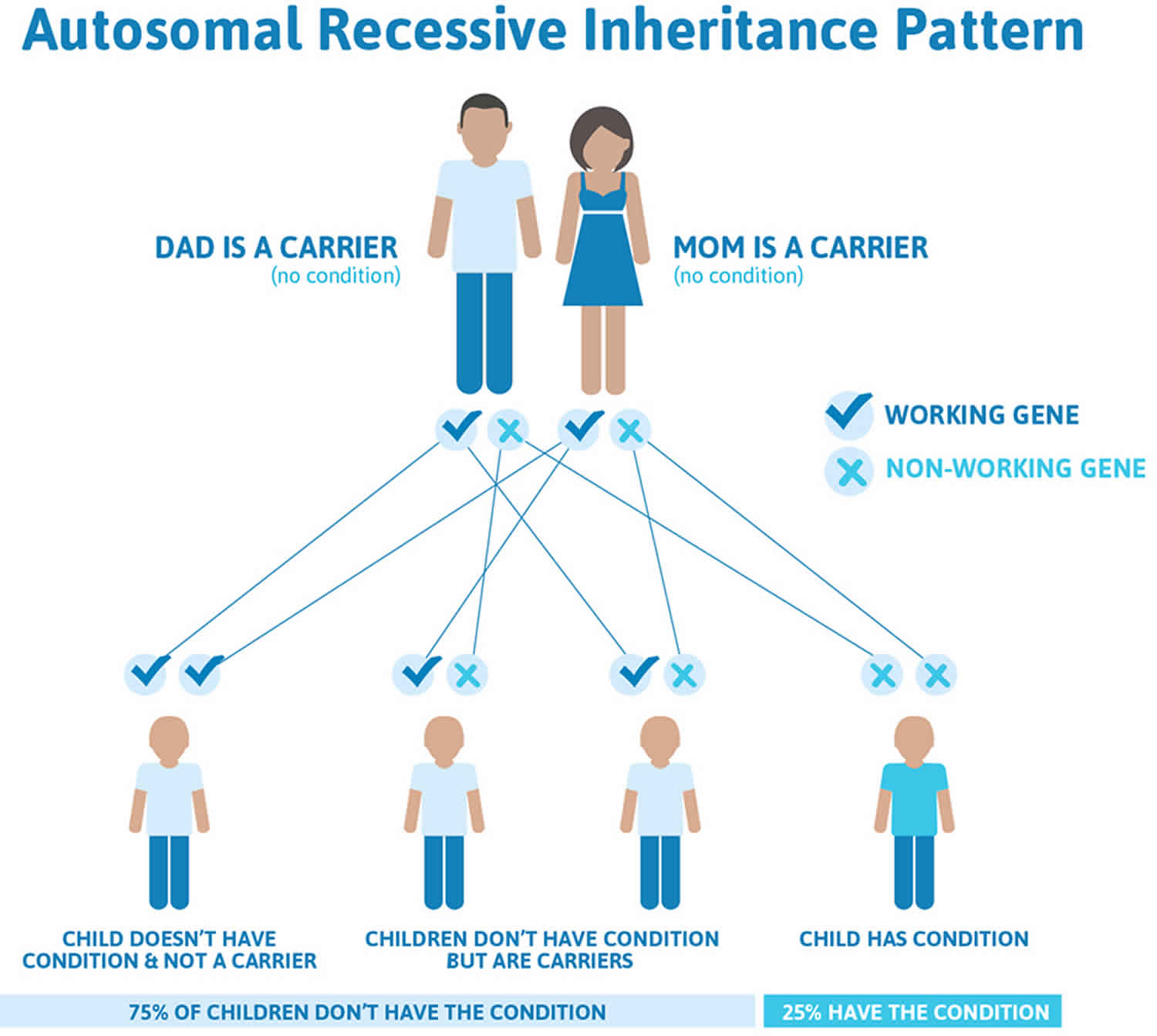
Diastrophic dysplasia is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 3 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Diastrophic dysplasia autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Diastrophic dysplasia symptoms
The symptoms and physical findings associated with diastrophic dysplasia may be extremely variable, differing in range and severity even among affected family members (kindreds). However, in all individuals with the disorder, there is abnormal development of bones and joints of the body (skeletal and joint dysplasia).
Symptoms of diastrophic dysplasia can include:
- Shortening of the limbs
- Scoliosis and increased lumbar lordosis with a prominent abdomen
- Hip dysplasia, in which the two hip joints are misaligned or crooked
- Deformities of the joints
- Foot deformities
- Hernia
- Cauliflower ear, a deformity of the cartilage of the ear
- Cleft palate, an opening or gap in the roof of the mouth
- Hitchhiker’s thumb, where the thumb sticks out at a right angle to the hand, looking like the child is trying to hitch a ride
- Brachydactyly, in which one or more of the fingers is abnormally short
During normal development before birth (embryonic and fetal development) as well as development during early childhood, cartilage in many areas of the body is gradually replaced by bone (ossification). In addition, a layer of cartilage (epiphyseal cartilage [growth plate]) separates the shafts (diaphyses) of long bones (e.g., bones of the arms and legs) from their ends (epiphyses), allowing long bones to grow until the cartilage is no longer present. In those affected by diastrophic dysplasia, however, there is delayed growth before and after birth (prenatal and postnatal growth retardation), the development of the ends of the long bones (epiphyses) is irregular, and the ossification of the epiphyses is delayed. Thus, affected newborns and children typically have markedly short, bowed arms and legs and short stature (short-limbed dwarfism). In addition, in such cases, growth failure is typically progressive, in part due to absence of the “growth spurt” that usually occurs during puberty. The severity of such growth failure may vary greatly from case to case, including among affected siblings.
Due to abnormalities of skeletal development, infants and children with diastrophic dysplasia also have additional distinctive malformations of bones of the hands, feet, and other areas of the body. For example, the first bone within the body of each hand (first metacarpals) may be unusually small, short, and “oval shaped.” As a result, the thumbs deviate away (abduction) from the body (“hitchhiker thumbs”). In addition, other fingers may be abnormally short (brachydactyly) and joints between particular bones of the fingers (proximal interphalangeal joints) may become fused (symphalangism), causing limited flexion and restricted movement (reduced mobility) of the finger joints. In some cases, bones of the wrists may also be malformed due to premature ossification.
Infants with the disorder also typically have severe foot deformities (talipes or “clubfeet”) due to abnormal fusion and deviation of bones within the body of each foot (metatarsals). In most cases, the heels turn outward (talipes valgus) while the fore part of each foot deviates inward (metatarsus adductus). In other infants, the soles of the feet may be flexed (talipes equinus) and, in some cases, the heels may also turn inward (talipes equinovarus). The great toes, like the thumbs, may also deviate away (abduction) from the body.
In addition to having limited flexion of finger joints, many affected infants and children also experience partial dislocation (subluxation) and/or complete dislocation of particular joints of the body. For example, in many cases, dislocations of the knees and hips occur upon weightbearing. Affected individuals may also have abnormally loose and/or stiff joints; experience limited extension of joints at the elbows and/or knees; and/or develop permanent flexion and immobilization (contracture) of certain joints (e.g., knees). Due to joint and bone abnormalities such as those affecting the feet, many individuals with diastrophic dysplasia have a tendency to walk on tiptoe. In addition, affected individuals may be predisposed to degenerative changes (osteoarthrosis) of particular joints (e.g. of the hips), resulting in pain with use of the joint, tenderness, stiffness, and, in some cases, deformity.
Many infants with diastrophic dysplasia also have abnormalities of bones within the spinal column (vertebrae). For example, in most affected infants, there may be incomplete closure of vertebrae (spina bifida occulta) within the neck area and the upper portion of the back (lower cervical and upper thoracic vertebrae) and/or abnormal narrowing of portions of the vertebrae of the lower back (interpedicular narrowing in lumbar vertebrae). During the first year of life, some infants may begin to develop progressive abnormal sideways curvature of the spine (scoliosis). In addition, during adolescence, individuals with diastrophic dysplasia may also develop abnormal front-to-back curvature of the spine (kyphosis), particularly affecting vertebrae of the neck region (cervical vertebrae). In severe cases, progressive kyphosis may result in difficulties breathing (respiratory distress). Some individuals with the disorder may also be prone to experiencing partial dislocation of joints between the central areas (bodies) of cervical vertebrae (cervical subluxation), potentially resulting in compression of the spinal cord. (This cylindrical structure of nerve tissue extends from the lower portion of the brain and is located inside the central canal within the spinal column [spinal cavity].) Such spinal cord injury may result in muscle weakness (paresis) or paralysis and/or life-threatening complications.
Most newborns with diastrophic dysplasia also have or develop fluid-filled sacs (cysts) within the outer, visible portions of the ears (pinnae). Within approximately two to five weeks after birth, the pinnae become swollen and inflamed. When such swelling and inflammation subside, the pinnae remain unusually thick, hard, and abnormal in shape. The abnormal areas of tissue (lesions) may gradually accumulate deposits of calcium salts (calcification) and eventually be replaced by bone (ossification). Although affected infants may experience associated abnormal narrowing (stenosis) of the external ear canal (external auditory canal), hearing is usually normal. However, according to reports in the literature, other affected infants and children may experience hearing impairment due to such auditory canal stenosis or abnormal fusion or absence of the three tiny bones (auditory ossicles) in the middle ear that conduct sound to the inner ear.
Some infants with diastrophic dysplasia also have characteristic malformations of the head and facial (craniofacial) area, such as an unusually high, prominent forehead; abnormal smallness of the jaws (micrognathia); and/or a broad, highly arched roof of the mouth (palate) or incomplete closure of the palate (cleft palate). Cleft palate has been reported to occur in anywhere from 25 to 60% of affected infants, and may cause difficulties with feeding and/or breathing. In addition, in some infants with diastrophic dysplasia, abnormalities of supportive connective tissue (cartilage) within the windpipe (trachea), voice box (larynx), and air passages in the lungs (bronchi) may cause abnormal narrowing (e.g., laryngotracheal stenosis) and collapse of such airways. In such cases, life-threatening complications such as respiratory obstruction and difficulties breathing (respiratory distress) may result. However, in many cases nasal speech (hyponasality) occurs as a result of the abnormally shaped vocal tract.
Approximately one third of infants and children with diastrophic dysplasia also have dental abnormalities, such as abnormally small teeth and dental crowding. In addition, in some cases, affected infants may have benign, reddish purple growths in the midportion of the face (midline frontal hemangioma) due to an abnormal distribution of tiny blood vessels (capillaries). Some individuals with the disorder may also have additional symptoms and physical findings.
Diastrophic dysplasia diagnosis
In some families with a previous history of diastrophic dysplasia, it is possible that the disorder may be detected before birth (prenatally) during early pregnancy (e.g., first trimester) based upon the results of specialized genetic (i.e., DNA marker) testing. In addition, in some cases, the disorder may be detected during mid pregnancy (e.g., second trimester) through fetal ultrasonography, a specialized imaging technique in which sound waves are used to create an image of the developing fetus. In such cases, diagnosis is most easily established when a clear family history is present. During fetal ultrasonography, a diagnosis of diastrophic dysplasia may be considered due to detection of certain characteristic findings, such as marked shortening of bones of the fingers (phalanges), arms, and legs; abnormal deviation (abduction) of the thumbs (“hitchhiker thumbs”) and great toes; severe deformities of both feet (talipes or “clubfeet”); and/or other findings.
In most cases, diastrophic dysplasia is diagnosed and/or confirmed at birth based upon a thorough clinical evaluation, identification of characteristic physical findings, and a variety of specializing tests, such as advanced imaging techniques.
The diagnosis of diastrophic dysplasia rests on a combination of clinical, radiologic, and histopathologic features. Diagnostic evaluation for diastrophic dysplasia begins with a thorough medical history and physical examination of your child. The diagnosis is confirmed by molecular genetic testing of SLC26A2, the only gene in which pathogenic variants are known to cause diastrophic dysplasia 9). Biochemical studies of fibroblasts and/or chondrocytes may be used in the rare instances in which molecular genetic testing fails to identify SLC26A2 pathogenic variants.
Doctors use a variety of diagnostic tests to diagnose diastrophic dysplasia and possible complications, including:
- X-rays, which produce images of bones.
- Magnetic resonance imaging (MRI), which uses a combination of large magnets, radiofrequencies and a computer to produce detailed images of organs and structures within the body.
- Computed tomography (CT) scan, which uses a combination of X-rays and computer technology to produce cross-sectional images (“slices”) of the body.
- EOS imaging, an imaging technology which creates 3-dimensional models from two planar images. Unlike a CT scan, EOS images are taken while the child is in an upright or standing position, enabling improved diagnosis due to weight-bearing positioning.
- Blood tests, which can help determine drug usage and effectiveness, biochemical diseases and organ function.
- Genetic testing, in which a sample of your child’s saliva is used to identify your child’s DNA.
- Radioisotope bone scan, which can help locate areas of abnormal growth.
- Arthrography, which uses colored dye injected into a joint — most commonly the shoulder, hip, knee, elbow or wrist — and X-ray images are taken to identify any problems.
Diastrophic dysplasia treatment
Every child’s condition is different, so treatment is determined on a case-by-case basis. The treatment of diastrophic dysplasia is directed toward the specific symptoms that are apparent in each individual. Treatment for diastrophic dysplasia is multi-pronged because the condition affects several body systems. Treatment may require the coordinated efforts of a team of specialists who may need to work together to systematically and comprehensively plan an affected child’s treatment. Such specialists may include pediatricians; physicians who diagnose and treat abnormalities of the skeleton, joints, muscles, and related tissues (orthopedists); surgeons; physical therapists; dental specialists (orthodontists); specialists who assess and treat hearing problems (audiologists); and/or other health care professionals.
In some cases, careful monitoring may be all that is required. Physicians may carefully monitor affected infants to ensure prompt detection and appropriate preventive or corrective treatment of respiratory obstruction and distress that may result due to certain abnormalities potentially associated with the disorder (e.g., laryngotracheal stenosis). In addition, special supportive measures may be used to help ensure an appropriate intake of nutrients in infants who experience feeding difficulties due to cleft palate. In some cases, surgical procedures may be performed to correct malformations resulting in breathing and/or feeding difficulties. The specific procedures performed will depend upon the location, severity, and combination of such anatomical abnormalities.
In other cases, nonsurgical or surgical treatments may be needed to address specific aspects of the condition.
For example, if your child has scoliosis, a team of orthopaedic surgeons will consider the severity of the curve, where it occurs in the spine, and your child’s age and stage of growth, before determining the best course of action. Treatment may include nonsurgical options such as bracing and physical therapy, or surgical options such as spinal fusion or implanting growing rods to stabilize your child’s spine as she continues to grow.
In affected children with dental abnormalities, braces (orthodontics), dental surgery, and/or other corrective procedures may be undertaken to correct such malformations. Steroid injections and/or other measures may also be used to help decrease the ear deformity that often affects infants with the disorder.
Many children with diastrophic dysplasia are also diagnosed with a variety of orthopaedic conditions including: hip dysplasia, and hand and foot anomalies. In some cases, these conditions are present at birth and can be treated when the child is young.
For example, a child with hitchhiker’s thumb will likely need hand surgery; while a child with a bowed leg may only need a long-leg cast.
In other cases the complication from diastrophic dysplasia may only become evident — or problematic — as your child grows. This is often true for spinal deformities such as scoliosis and hip conditions.
In some cases, physical therapy in combination with surgical and supportive measures may be helpful in improving an affected individual’s ability to walk and perform other movements (mobility). According to the medical literature, although the foot deformities (i.e., talipes or clubfeet) associated with the disorder may be resistant to treatment, early, persistent therapy may be helpful in achieving beneficial results. In addition, because particular skeletal changes associated with diastrophic dysplasia are progressive (e.g., kyphosis) and, in some cases, may lead to severe complications (e.g., respiratory distress, compression of the spine, potential paresis or paralysis), physicians may perform ongoing monitoring to ensure prompt detection of and appropriate preventive and/or corrective measures for such abnormalities.
Genetic counseling will be of benefit for affected individuals and their families. Other treatment for this disorder is symptomatic and supportive.
Follow-up care
Your child with diastrophic dysplasia should continue to be monitored by an orthopaedic physician into adulthood. Annual visits to follow the development of the spine and hips is appropriate.
If your child had spine surgery, he or she will need to see the orthopaedic surgeon about one to two weeks after surgery, then again at three and six months post-surgery. After that, annual monitoring by trained clinicians is strongly encouraged to ensure any problems are spotted and treated as soon as possible.
Additionally, physicians may recommend your child see several different specialists because so many body systems are involved in a diagnosis of diastrophic dysplasia.
For example, your child may see:
- An otolaryngologist or plastic surgeon for cleft palate
- A geneticist for individual or family counseling
- An orthopaedist for any bone- and muscle-related issues
- A pulmonologist for any breathing issues
- Physical therapists and occupational therapists to expand your child’s physical dexterity and skill
- Nutritionists to improve your child’s dietary health
During follow-up visits, X-rays and other diagnostic testing may be done. The goal of continued monitoring is to help spot any irregularities in growth or development and to address health issues as they develop.
Diastrophic dysplasia life expectancy
Children with diastrophic dysplasia can lead normal lives. They can hold good jobs, get married, have children and more.
If you have questions about how your child’s condition and any related health issues may affect your child’s prognosis or long-term goals, talk to your child’s healthcare provider.
References [ + ]




{kind=link}