



Childhood glaucoma
Childhood glaucoma also called congenital glaucoma, pediatric glaucoma or infantile glaucoma, is glaucoma that occurs in babies and young children. Childhood glaucoma is usually diagnosed within the first year of life. Glaucoma is a condition in which the normal fluid pressure inside the eyes (intraocular pressure or IOP) slowly rises as a result of the fluid aqueous humor – which normally flows in and out of the eye – not being able to drain properly from the anterior chamber of the eye resulting in increased pressure within the eye. Instead, the fluid collects and causes pressure damage to the optic nerve (a bundle of more than 1 million nerve fibers that connects the retina with the brain) and loss of vision.
Glaucoma is classified according to the age of onset. Glaucoma that begins before the child is 3 years old is called infantile or congenital (present at birth) glaucoma. Glaucoma that occurs in a child is called childhood glaucoma. Infantile glaucoma develops between the ages of 1-24 months. Glaucoma with onset after age 3 years is called juvenile glaucoma. Another way to classify glaucoma is to describe the structural abnormality or systemic condition which underlies the glaucoma.
Childhood glaucoma is a rare condition that may be inherited, caused by incorrect development of the eye’s drainage system before birth. This leads to increased intraocular pressure (IOP), which in turn damages the optic nerve. It is estimated that 1 in 5,000 to 10,000 children under 2 years of age will develop primary congenital/ primary infantile glaucoma. However, if a child has cataract surgery or one of the other conditions listed above, the incidence of glaucoma will be much higher. For example, 50% of patients with aniridia will develop glaucoma during their lifetime.
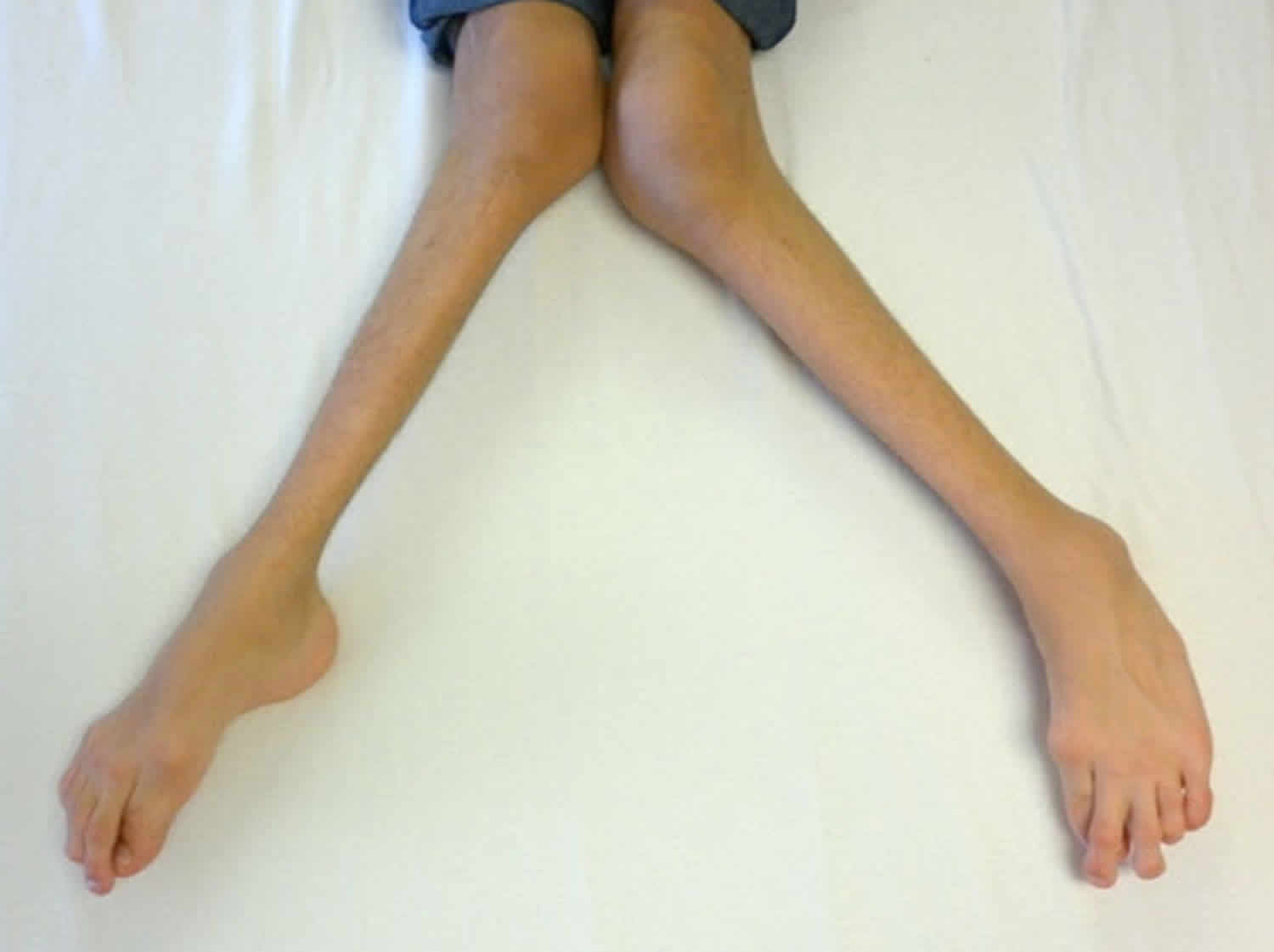
Symptoms of childhood glaucoma include enlarged eyes, cloudiness of the cornea, and photosensitivity (sensitivity to light). Your child needs to be assessed by an ophthalmologist. Ideally the child should be referred to an ophthalmologist with experience in managing childhood glaucoma. The nature of assessment undertaken varies with the age of the child. In children over 7 years the tests used are very similar to those used for adults, i.e. pressure measurement, visual field assessment and examination of the optic disc after dilatation of the pupil.
Both medication and surgery are required in some cases. In an uncomplicated case, surgery can often correct such structural defects.
The treatment for glaucoma in older children is generally medical (eye drops) initially and if these fail, surgery is considered. This is similar to the situation with older adults with glaucoma.
Medical treatments may involve the use of topical eye drops and oral medications. These treatments help to either increase the exit of fluid from the eye or decrease the production of fluid inside the eye. Each results in lower eye pressure.
There are two main types of surgical treatments: filtering surgery and laser surgery. Filtering surgery (also known as micro surgery) involves the use of small surgical tools to create a drainage canal in the eye. In contrast, laser surgery uses a small but powerful beam of light to make a small opening in the eye tissue. Most glaucoma surgery in children can be done safely as a day case without the need for overnight admission. If surgery is required within the first four to five weeks of life the infant may remain in overnight for monitoring after the anesthetic.
Operations such as trabeculectomy or drainage tubes are used. These procedures aim to create a controlled leak or “fistula” by which the aqueous can bypass the trabecular meshwork and escape to drain via the external blood system of the eye.As with adults anti-inflammatory and antibiotic drops are used post operatively. When trabeculectomy is performed in children an anti-metabolite such as 5-fluorouacil (5-FU for short) is very often used as children heal much more rapidly than adults.Laser trabeculoplasty is rarely used in the treatment of glaucoma in children of any age. A cyclo-destructive procedure, such as diode laser treatment of the ciliary body, is sometimes used in the treatment of aphakic glaucoma in children.
Despite timely and aggressive treatment, childhood glaucoma can still cause significant and permanent vision loss. Early diagnosis and treatment, as well as close monitoring are crucial for obtaining a long-term successful outcome.
Figure 1. Eye anatomy
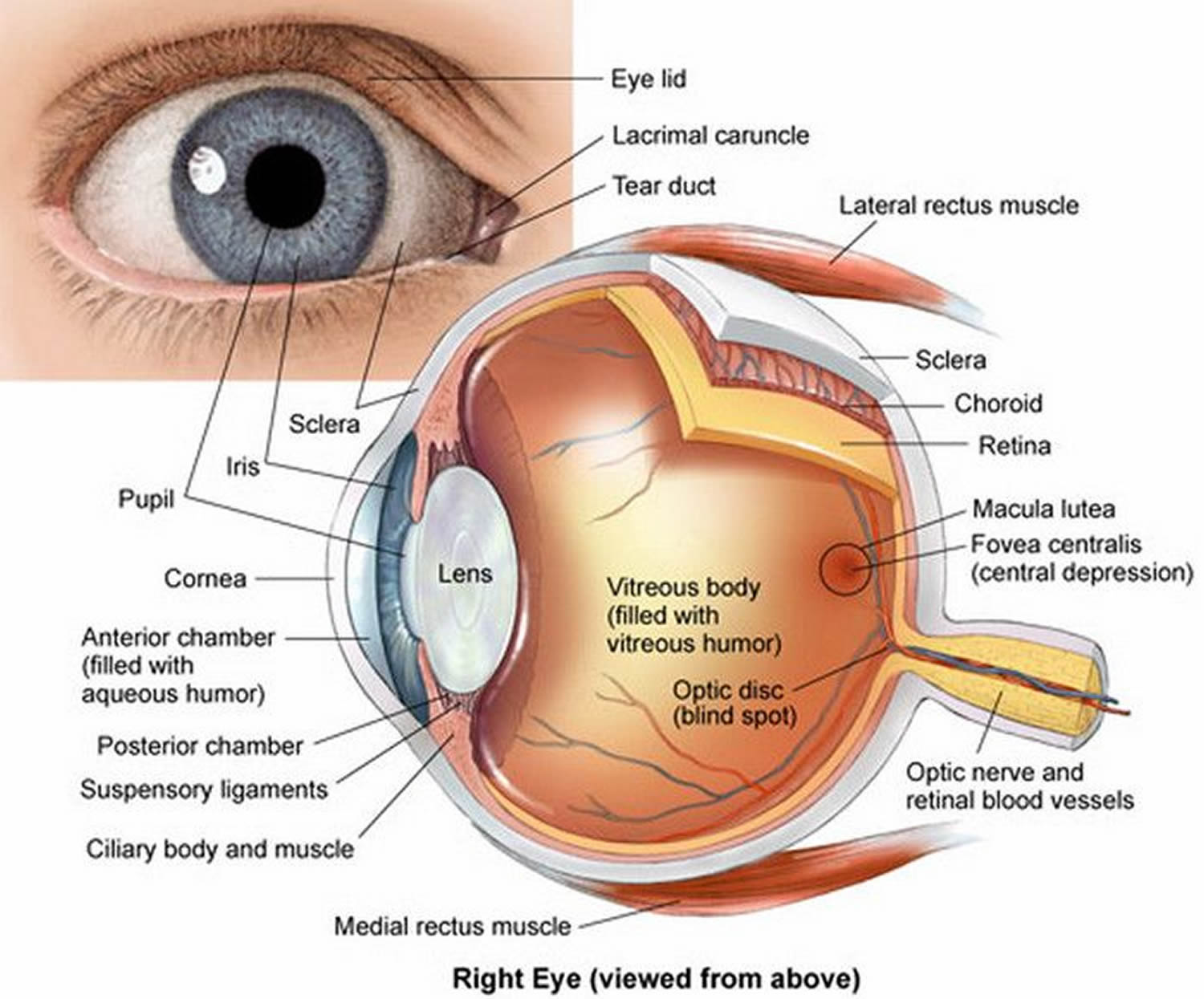
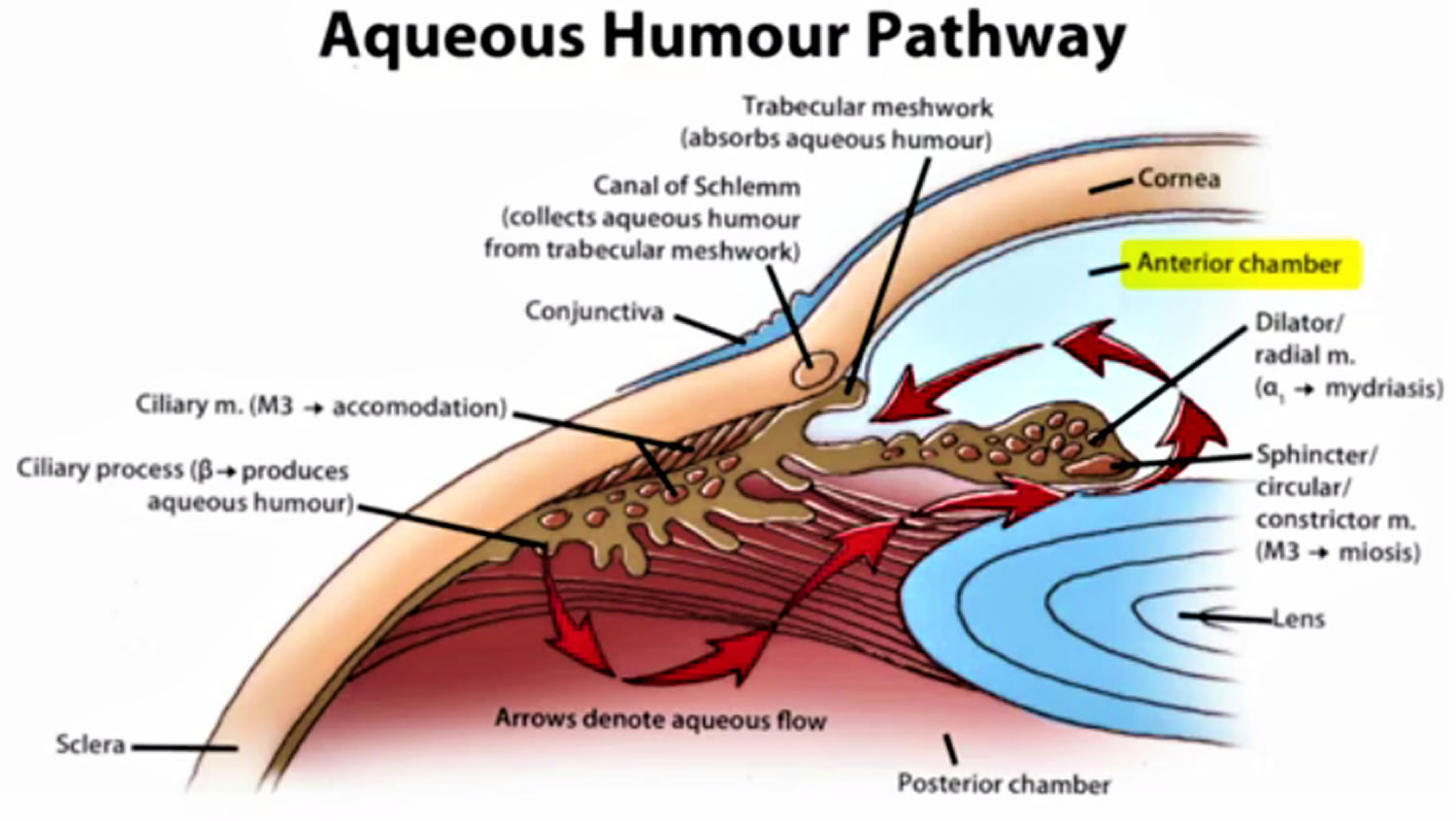
Figure 2. Normal aqueous outflow
How would a parent know if a child is suffering from glaucoma?
In keeping with the rest of an infant the immature eye is floppy and somewhat elastic. Thus early in life (that is before the second birthday) raised intraocular pressure will stretch the eye and actually cause it to increase in size (it expands in all directions/ rather like a balloon being inflated). Early medical writers termed this buphthalmos (ox eye) as this increase in size of the eye was thought to make the infant’s eye look like an ox’s eye.
The stretching of the eye has a number of harmful effects on the eye. As the eye enlarges the cornea increases in size. One of the many layers of the cornea, Descemet’s membrane, does not have much give and rather than stretch it will split as the eye enlarges. This splitting results in the cornea losing some of its clarity and becoming cloudy. This cloudiness of the cornea is the result of fluid entering the cornea from the anterior chamber via the splits in Descemet’s membrane and is known as corneal edema. Corneal edema causes discomfort and sensitivity to light and increased tear production.
Thus the cardinal features of infantile glaucoma that may be identified by a parent are photophobia (sensitivity to light), increased tearing with an enlarged and cloudy cornea.
What happens when glaucoma is suspected in a young child?
The child needs to be assessed by an ophthalmologist (eye specialist). Ideally the child should be referred to an ophthalmologist with experience in managing childhood glaucoma. Often an examination under anesthetic is required to adequately examine the child and confirm the diagnosis of glaucoma. The diagnosis is confirmed by the presence of typical corneal changes (enlargement, clouding and splits in Descemet’s membrane), raised intraocular pressure and optic disc cupping.
Will my child’s vision be impaired?
Severe loss of vision due to infantile glaucoma is fortunately rare. However, if glaucoma is not appropriately treated there is a risk of progressive visual impairment. Rarely does childhood glaucoma result in severe visual impairment but life-long follow-up is needed for all children after a diagnosis of glaucoma is made. Vision impairment is particularly seen if the onset of the glaucoma is at or before birth. Glasses are commonly required for myopia (short sightedness). This is due to the overall length of the eyeball being increased by the raised intraocular pressure. Photophobia may be a persistent problem if the splits in Descemet’s membrane are severe.
Therefore prompt recognition and timely treatment will improve the chance of a good vision outcome.
Will my child’s lifestyle need to alter in any way?
Most children with infantile glaucoma lead normal lives. Glasses may be required for focusing errors or photophobia. Adolescents often have difficulty accepting the need for long-term medication and regular medical review. Ensuring compliance with regular use of eye drops may be especially difficult. The small number of children with more severe visual impairment will require some degree of help at school.
Is childhood glaucoma hereditary?
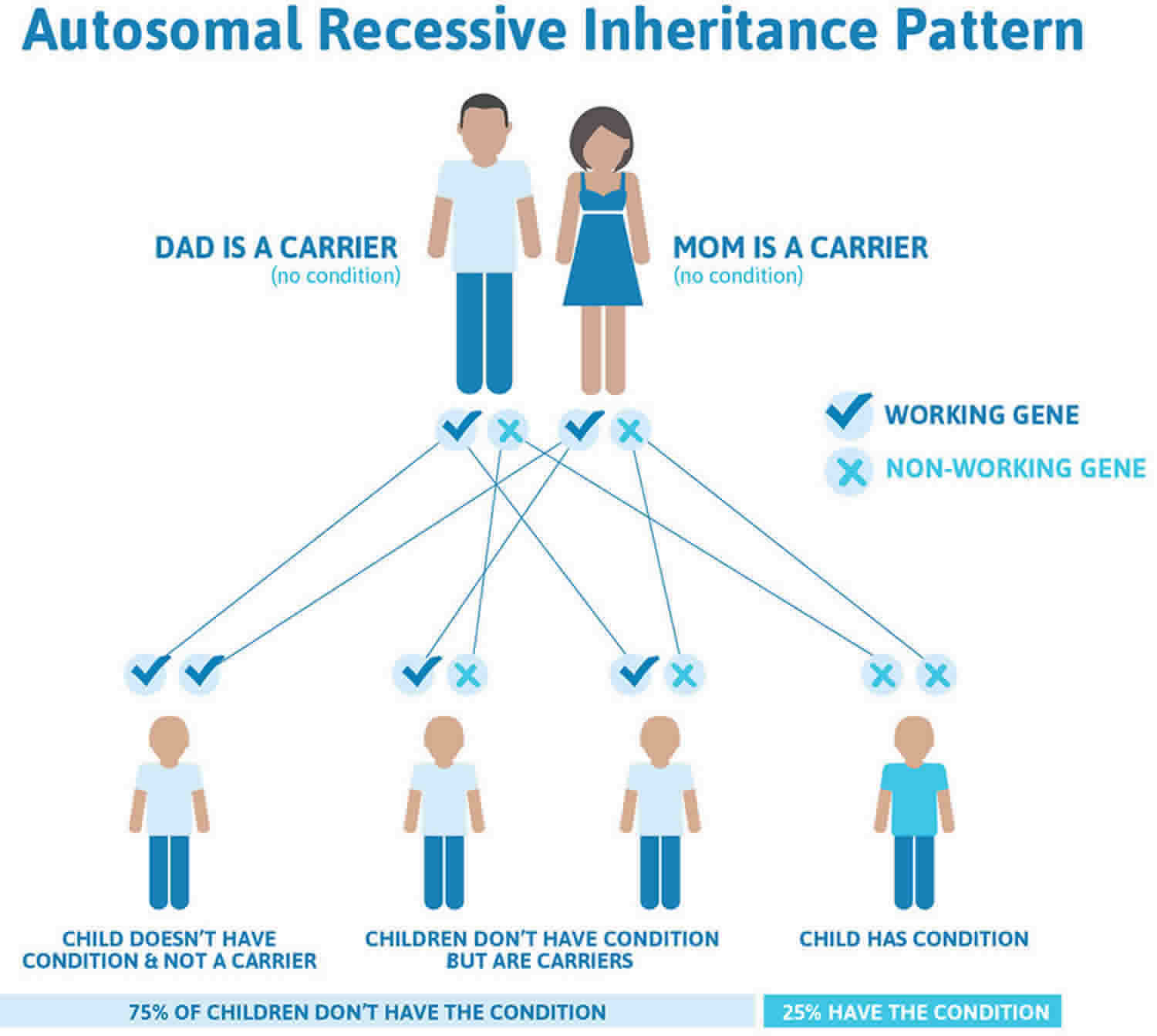
Some types of childhood glaucoma are hereditary. About 10% of primary congenital/infantile glaucoma cases are inherited. Recent research has identified some specific gene mutations linked to this disease; for which genetic testing and counseling for affected families is may be available.
Other conditions that cause secondary glaucoma can be inherited. For example, neurofibromatosis and aniridia are dominantly inherited and are passed on to the children of affected individuals approximately 50% of the time. The incidence of glaucoma that occurs in association with these conditions, however, is less predictable.
What are the chances of another baby of mine developing glaucoma?
The risk is not zero but it is quite low. Primary open angle glaucoma in adolescents may show a familial tendency just as in adult open angle glaucoma. Inherited juvenile open angle glaucoma is well recognized but very rare. This form of glaucoma is generally not detectable till the twenties rather than during later childhood.
Childhood glaucoma causes
Glaucoma occurs when the fluid drainage from the eye is blocked by abnormal development or injury to the drainage tissues, thus, resulting in an increase in the intraocular pressure, damage to the optic nerve, and loss of vision.
There are many causes of childhood glaucoma. It can be hereditary or it can be associated with other eye disorders. If glaucoma cannot be attributed to any other cause, it is classified as primary glaucoma. If glaucoma is a result of another eye disorder, eye injury, or other disease, it is classified as secondary glaucoma.
Approximately 20% of children with primary congenital glaucoma have a mutation in the CYP1B1 gene. If one child in a family has primary congenital glaucoma then the risk for a subsequent child to be affected is about5% and if there are two affected children in one family the risk for subsequent children is 25%. Data suggests that the risk for a parent with a diagnosis of primary congenital glaucoma of having an affected child is 2%.
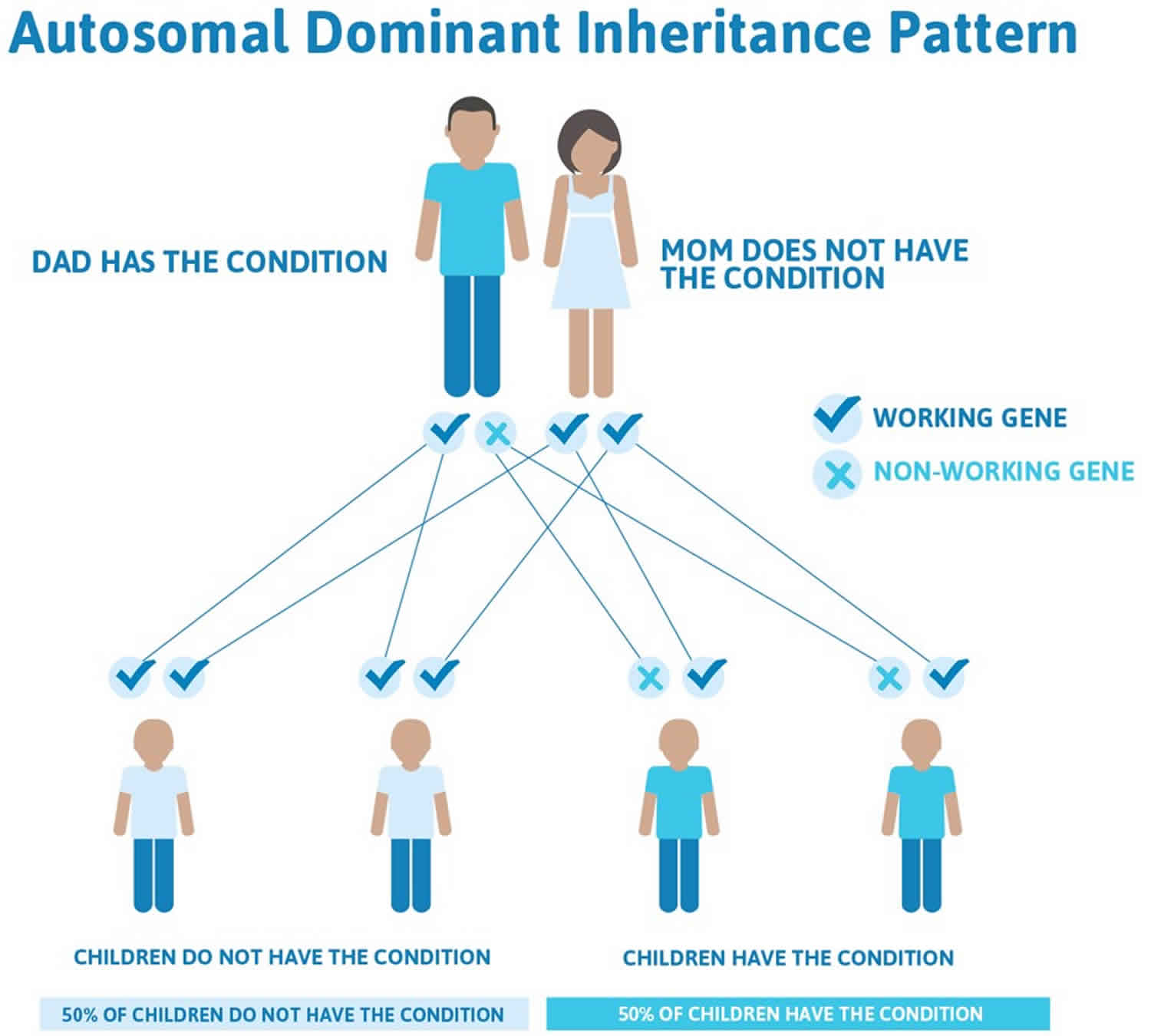
Most primary congenital glaucoma cases are sporadic (with no family history), however, about 10-40% are familial, with an autosomal recessive inheritance pattern and penetrance varying from 40-100% 1). Autosomal dominant inheritance has also been reported 2). Five loci have been identified by linkage analyses: GLC3A (located on choromosome 2p22-p21), GLC3B (1p36.2-p36.1), GLC3C (14q24.3), GLC3D (14q24.2-q24.3, not overlapping with GLC3C), and GLC3E (9p21) 3).
Thus far, a gene associated with primary congenital glaucoma has been identified in three of these five loci. Further details are below.
The GLC3A loci contains the cyctochrome P4501B1 (CYP1B1) gene, which was the first reported primary congenital glaucoma-causing gene. It codes for an enzyme that metabolizes compounds vital for the developing eye, and is expressed in fetal and adult neuroepithelium and ciliary body 4). Severe trabecular meshwork atrophy is seen in mouse models deficient of CYP1B1 5). In zebrafish, CYP1B1 has been found to indirectly affect neural crest migration to the anterior segment and angle by playing a role in ocular fissure closure 6). While the exact mechanism by which CYP1B1 mutations causes primary congenital glaucoma is unknown, scientists know that CYP1B1 mutations are associated with 15-20% of primary congenital glaucoma cases in Japan and the United States, and all cases in Slovakia Roma 7).
The GLC3D locus contains latent transforming growth factor beta binding protein 2 (LTBP2). LTBP2 is expressed in trabecular meshwork and ciliary processes however its role in the eye is unknown 8). In nonocular tissues, it is involved in tissue repair and cell adhesion 9). LTBP2 null mutations have been reported in consanguineous Iranian and Pakistani families, and Slovakian Roma with primary congenital glaucoma. Homozygosity for a variant of an LTBP2 mutation was associated with worse outcomes even while undergoing more surgical interventions 10).
GLC3E contains the tunica interna endothelial cell kinase (TEK, also known as TIE2) gene. The angiopoietin/TEK (ANGPT/TEK) signaling pathway is required in Schlemm canal development, and includes 3 ligands (ANGPT1, ANGPT2, and ANGPT4) and 2 receptors (TEK and TIE). TEK-knockout mice can have no Schlemm canal and TEK-hemizygous mice have a severely underdeveloped Schlemm canal 11). Additionally, ANGPT1 mutations have been identified in a few primary congenital glaucoma patients who do not have other known primary congenital glaucoma-causing genes, revealing another member of the ANGPT/TEK signaling pathway that may be a cause of primary congenital glaucoma 12).
Lastly, though not associated with the above loci, but originally associated with juvenile open angle glaucoma and primary open angle glaucoma, the myocilin/trabecular meshwork-induced glucocorticoid response protein (MYOC) gene on chromosome 1q24 may also explain a small proportion of primary congenital glaucoma cases, up to 5.5% 13).
Primary congenital glaucoma patients may have one or more genes affected, and the previously discussed genes may regulate or interact with each other 14). CYP1B1 may play a role as a modifier gene for MYOC expression 15) and a digenic mode of inheritance has been considered for CYP1B1 and MYOC 16) and CYP1B1 and TEK 17).
Currently, the chance of identifying a genetic cause is 40% when genetic testing is done 18).
Infantile and childhood glaucoma may be associated with other abnormalities in the eye. The most common of these is a history of having had cataract surgery as an infant, this is called “aphakic glaucoma”. Other eye abnormalities that can be associated with glaucoma are the anterior segment dysgenesis group of disorders which includes Axenfeld-Reiger syndrome. Glaucoma can occur in association with other systemic abnormalities such as the Sturge-Weber syndrome and rubella (German measles) embryopathy. Juvenile arthritis may cause inflammation in the eye (uveitis) that may be complicated by the development of glaucoma. Primary open angle glaucoma does occur rarely in older children and adolescents.
Childhood glaucoma symptoms
Glaucoma is rare in children, as compared to the adult. However, when it does occur, the symptoms may not be as obvious in children. Many children are diagnosed before they are 6 months old. Glaucoma can affect one eye or both.
The most common symptoms of congenital/infantile glaucoma are excessive tearing, light sensitivity and a large, cloudy cornea (the normally clear front surface of the eye) which can cause the iris (colored part of the eye) to appear dull. Excessive tearing accompanied by mattering/discharge in a child is usually not caused by glaucoma but instead is the result of congenital nasolacrimal duct obstruction (blocked tear duct).
Juvenile glaucoma tends to develop without any obvious symptoms, similar to adult glaucoma. Patients with juvenile glaucoma often have a positive family history. On exam the eye pressure will typically be elevated and there may be signs of optic nerve cupping (enlargement of the center “cup” portion of the optic nerve).
The following are the most common symptoms of childhood glaucoma. However, each child may experience symptoms differently. Symptoms may include:
- excessive tearing
- light sensitivity (photophobia)
- closure of one or both eyes in the light
- cloudy, enlarged cornea (large eye)
- one eye may be larger than the other
- vision loss
If the eye pressure increases rapidly, there may be pain and discomfort. Parents may notice that the child becomes irritable, fussy, and develops a poor appetite. Early detection and diagnosis is very important to prevent loss of vision. The symptoms of glaucoma may resemble other eye problems or medical conditions. Always consult your child’s physician for a diagnosis.
Childhood glaucoma diagnosis
In addition to a complete medical history and eye examination of your child, diagnostic procedures for childhood glaucoma may include:
- visual acuity test – the common eye chart test (with letters and images), which measures vision ability at various distances.
- pupil dilation – the pupil is widened with eyedrops to allow a close-up examination of the eye’s retina and optic nerve.
- visual field – a test to measure a child’s side (peripheral) vision. Lost peripheral vision may be an indication of glaucoma.
- tonometry – a standard test to determine the fluid pressure inside the eye.
Younger children may be examined with hand-held instruments, whereas older children are often examined with standard equipment that is used with adults. An eye examination can be difficult for a child. It is important that parents encourage cooperation. At times, the child may have to be examined under anesthesia, especially young children, in order to examine the eye and the fluid drainage system, and to determine the appropriate treatment.
Childhood glaucoma treatment
Specific treatment for glaucoma will be determined by your child’s opthalmologist (eye specialist) based on:
- your child’s age, overall health, and medical history
- extent of the disease
- your child’s tolerance for specific medications, procedures, or therapies
- expectations for the course of the disease
- your opinion or preference
It is important for treatment of childhood glaucoma to start as early as possible. Childhood glaucoma is treated by lowering the intraocular pressure (IOP) via medical and/or surgical means. Treatment may include:
- Medications: Some medications cause the eye to produce less fluid, while others lower pressure by helping fluid drain from the eye.
- Conventional surgery: The purpose of conventional surgery is to create a new opening for fluid to leave the eye. Surgical procedures are performed by using microsurgery or lasers. The purpose of surgery is to create an opening for fluid to leave the eye. Surgical procedures used to treat glaucoma in children include the following:
- Trabeculotomy and goniotomy: A surgical opening is made into the drainage area of the eye (known as the trabecular meshwork drainage system), therefore establishing a more normal anterior chamber angle that allows the fluid to drain more freely, lowering the intraocular pressure (IOP). A goniotomy is an internal trabeculotomy procedure that is used in congenital glaucoma.
- Trabeculectomy: A surgical procedure that involves the removal of part of the trabecular meshwork drainage system, allowing the fluid to drain from the eye.
- Iridotomy: In this procedure, a small hole is made through the iris – the colored part of the eye – to allow fluid to flow more freely in the eye. The surgeon may use a laser to create this hole (laser iridotomy).
- Cyclophotocoagulation: A procedure that uses a laser beam to freeze selected areas of the ciliary body – the part of the eye that produces aqueous humor – to reduce the production of fluid. This type of surgery may be performed with severe cases of childhood glaucoma.
Both medications and surgery have been successfully used to treat childhood glaucoma. Most cases of primary childhood glaucoma are treated with surgery.
Initial treatment may be eye drops or medication by mouth to lower the pressure in the eye. Over the long-term medications have a significant risk of complication in young children and compliance with medical therapy is an even greater problem with young patients than it is with older ones. Surgery is usually required and has a very high success rate.
Childhood glaucoma is the result of blockage of aqueous (fluid) drainage at the trabecular meshwork of the anterior chamber angle. Operations aim to restore the more normal drainage of aqueous. The two most common operations are goniotomy and trabeculotomy. Both involve opening up the tissue in the angle to enable the aqueous to escape more easily from the eye and thus lower the pressure. All surgery for childhood glaucoma is done under a general anesthetic. It is not uncommon for more than one operation to be needed to completely control the raised pressure of infantile glaucoma.
In some instances operations more often used with adult glaucoma such as trabeculectomy or Molteno tubes are used. These procedures aim to create a controlled leak or “fistula” by which the aqueous can bypass the trabecular meshwork and escape from the eye. As with adults anti-inflammatory and antibiotic drops are used post operatively. When trabeculectomy is performed in children an antimetabolite such as 5-fluorouacil (5-FU for short) is very often used as children heal much more rapidly than adults.
Many children with pediatric glaucoma develop myopia (nearsightedness) and require glasses. Also, amblyopia (“lazy eye”) and strabismus (misalignment of the eyes) occur more frequently and may require treatment with patching or surgery.
Follow up
Regular and life long follow up will be required. When the child is quite young it may be necessary for periodic examinations under anaesthetic. As well as monitoring for raised intraocular pressure the follow up will involve monitoring of the development of vision, determine the need for glasses and when older assess any damage to peripheral visual field.
References [ + ]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}