



Pediatric lupus
Pediatric lupus also known as pediatric systemic lupus erythematosus (pSLE) is a disease in which the immune system is overactive and does not function properly. The immune system attacks the body and creates inflammation in the skin, joints, kidneys, lungs, nervous system, and other organs of the body. People with lupus can have times of very active disease, called a flare, and times where the disease is mostly quiet, called remission.
When SLE occurs in children doctors tend to call it pediatric systemic lupus erythematosus (pSLE) because it typically hits kids harder than adults and carries extra health risks, since children have more years to accrue organ damage compared with adults.
About 20 percent of people with lupus or SLE developed the disease before 20 years of age. It is rare to get lupus before age 5 years. In the U.S., lupus affects an estimated 5,000 to 10,000 youngsters. Lupus is more common in females and in certain ethnic groups, including African-American, Hispanic, South and Southeast Asian and North American First Nations populations.
Pediatric lupus key facts
- Lupus is a chronic disease, with flares and remissions.
- Lupus is not contagious and it cannot be prevented.
- Lupus can affect many different areas of the body.
- Children with lupus are more likely to have problems with vital organ systems — most critically, the kidneys and the central nervous system (brain and spinal cord)
- Children with lupus develop damage from their disease more quickly
- Children with lupus have a higher “burden of disease” over their lifetime (meaning that the earlier their lupus begins, the more years they spend living with it)
- Treatment is different for each child; each child is unique, as is each treatment plan.
- Lupus and several medications used for lupus suppress the immune system. Work with your rheumatology team to learn about lupus and find the best treatment plan to control it.
- Becoming more involved in your care will help as you grow with this illness to make choices and transition into adulthood.
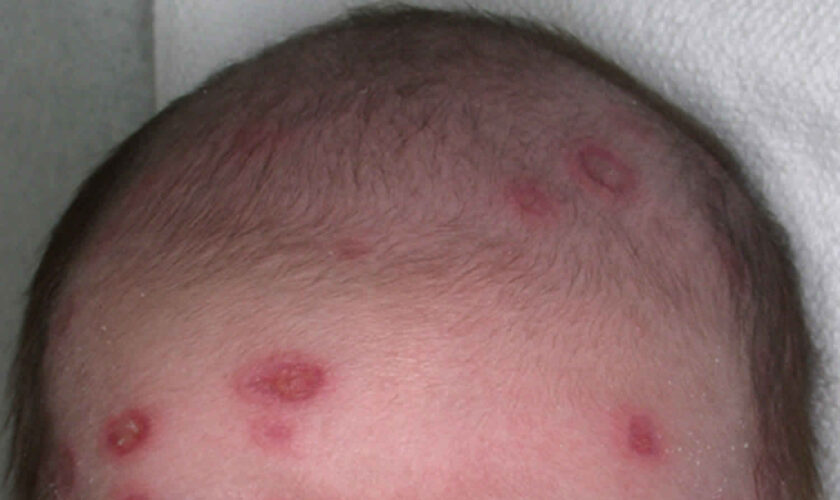
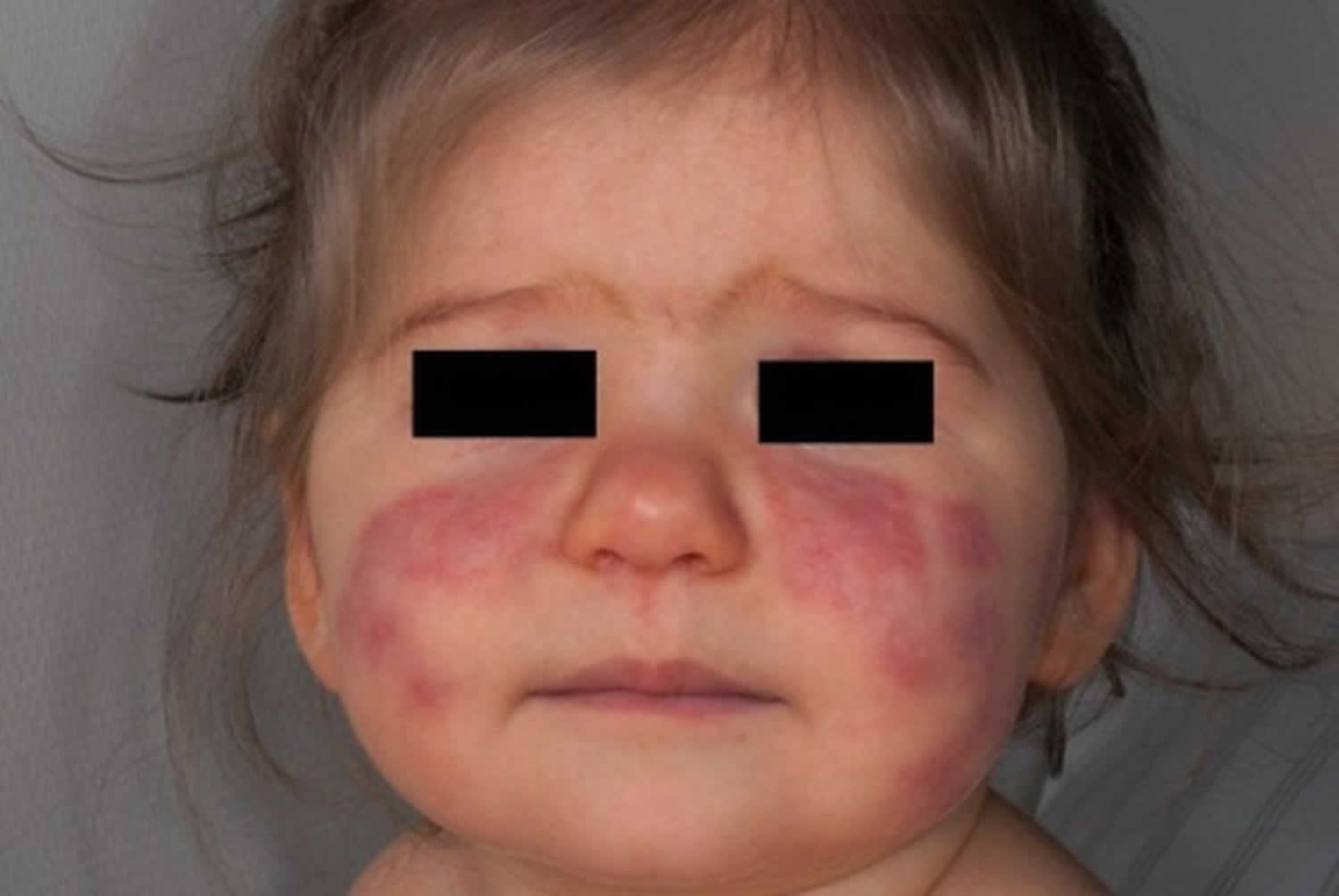
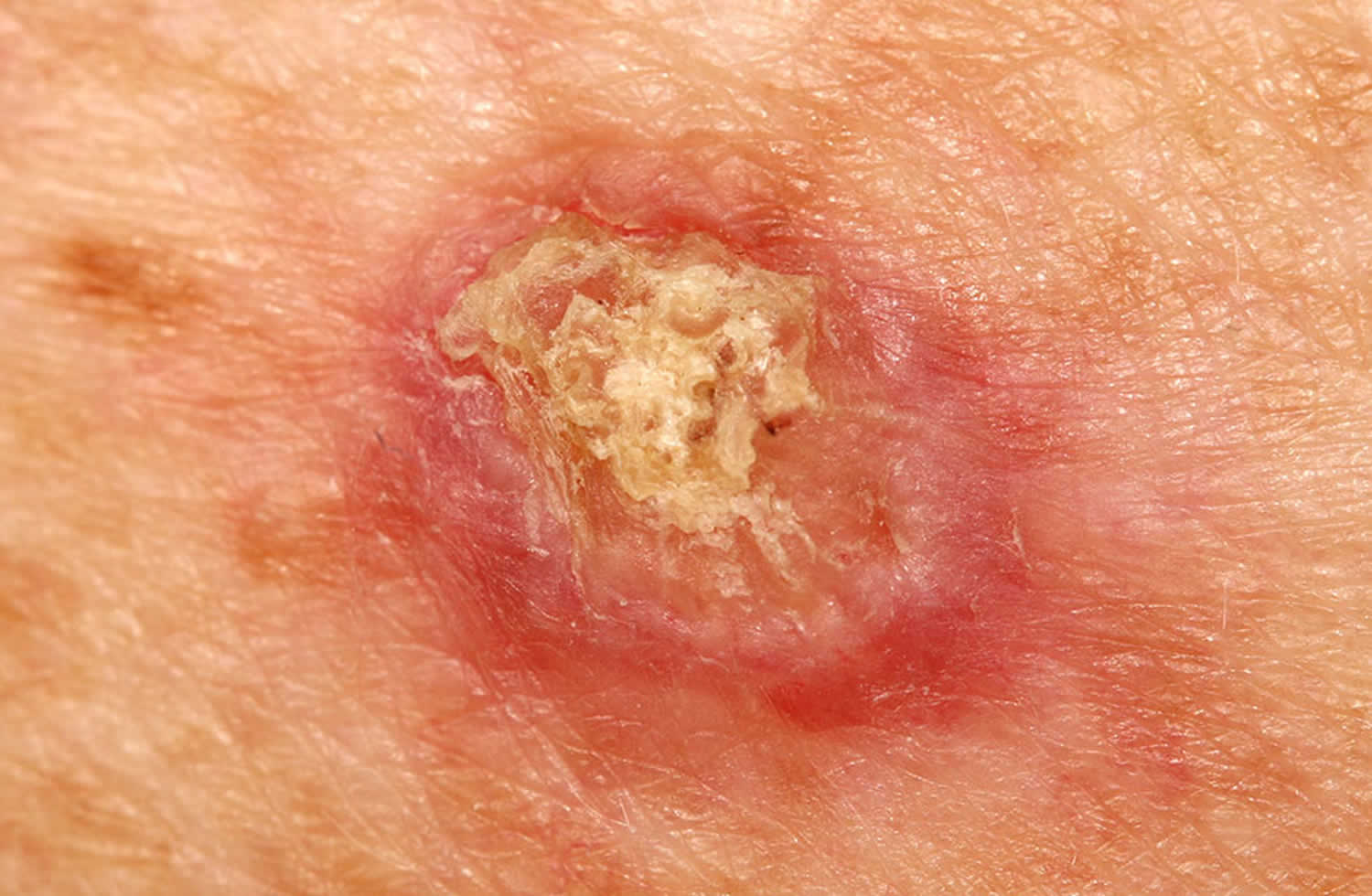
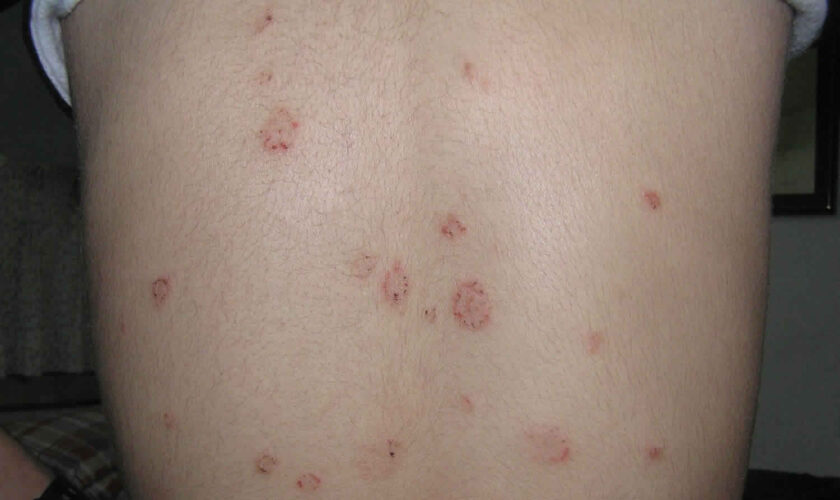
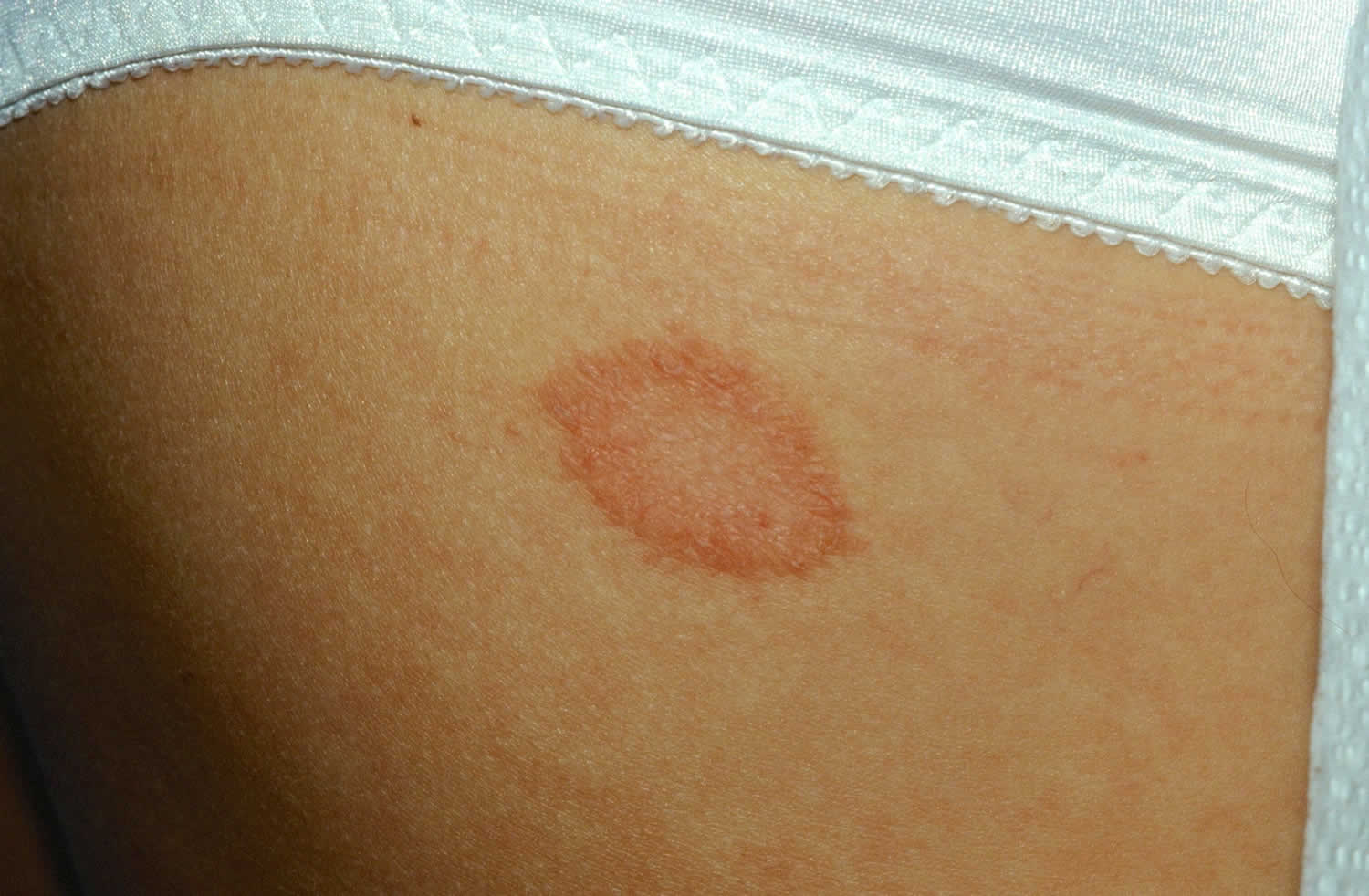
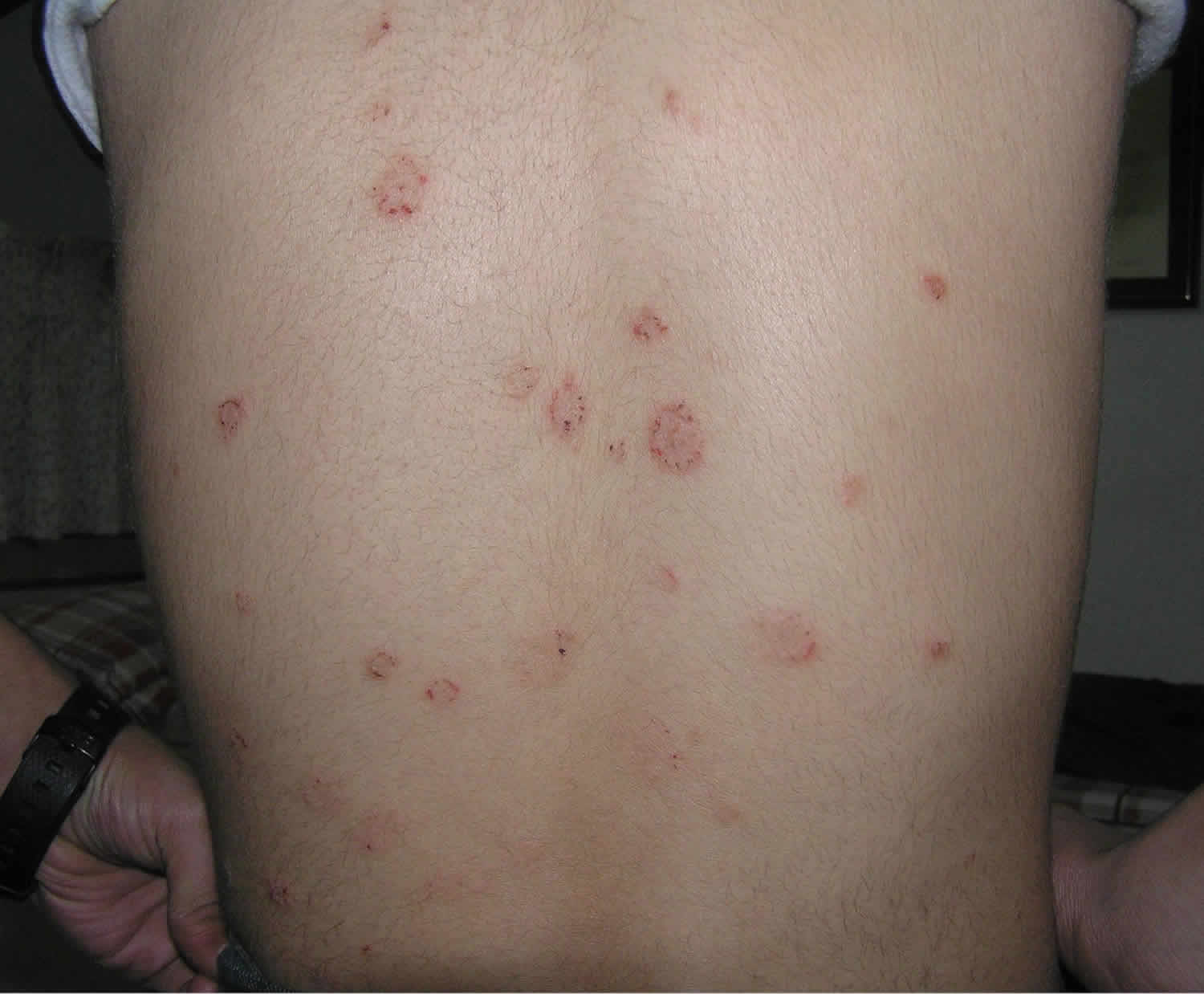
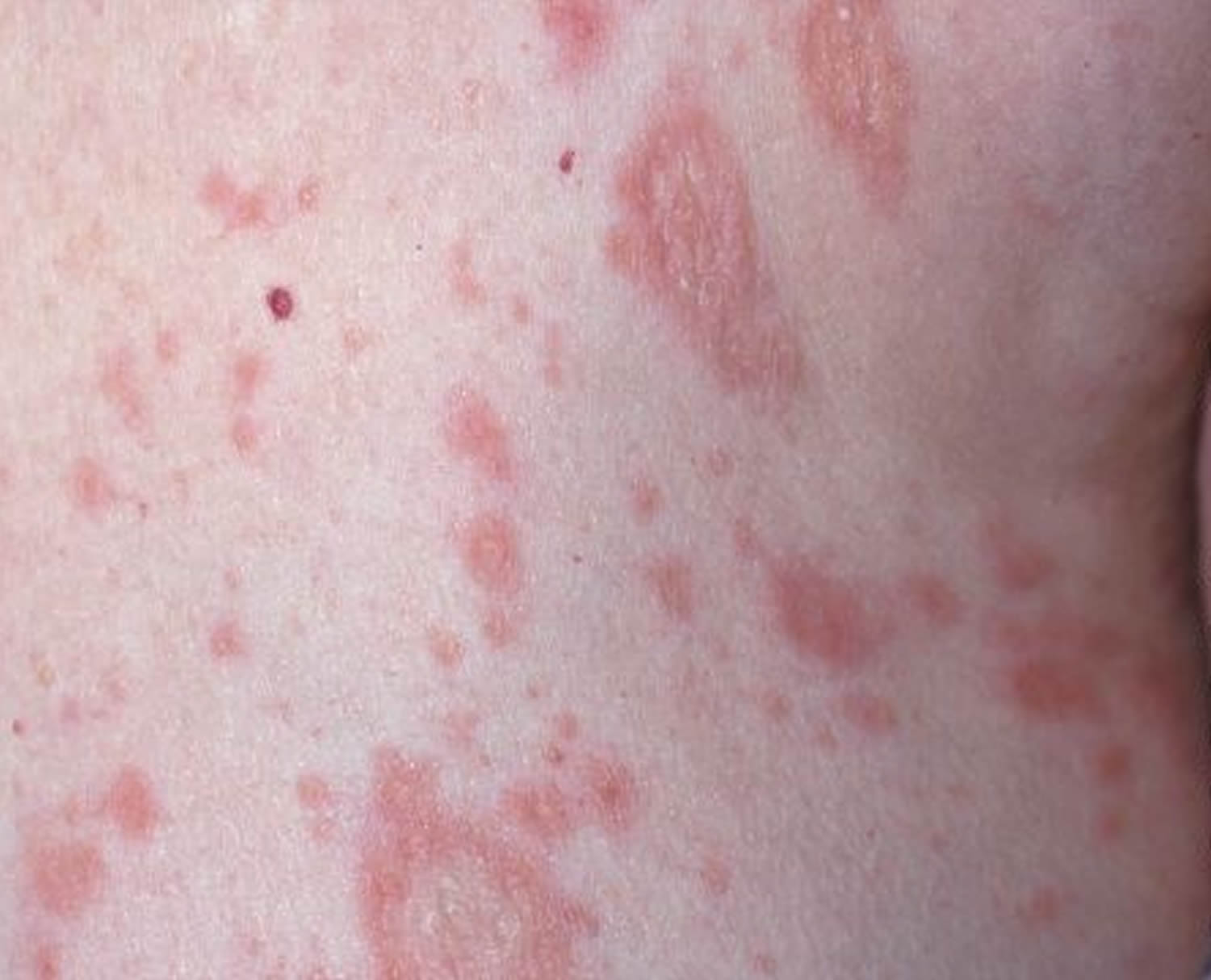
Figure 1. Lupus rash in children
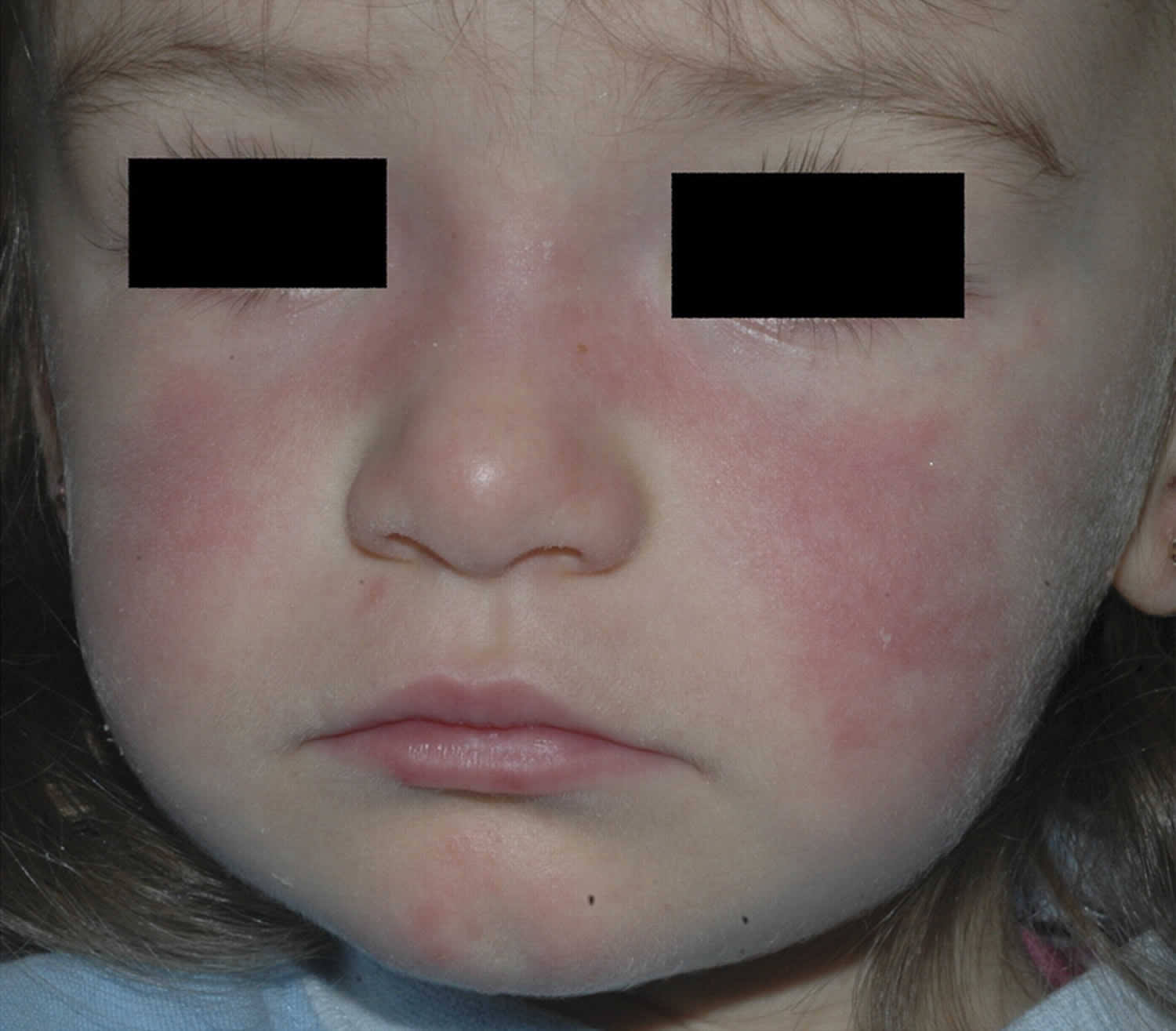
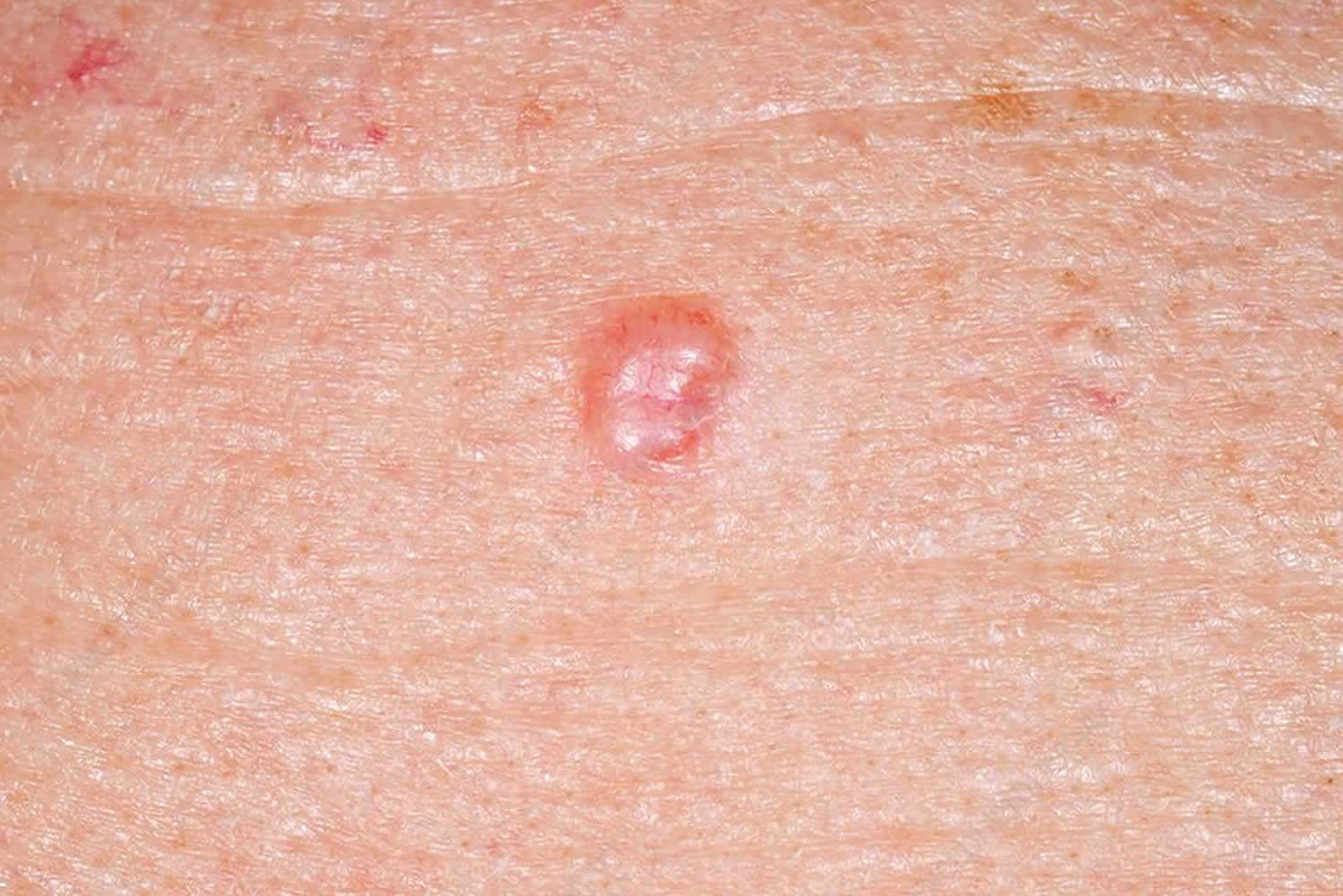
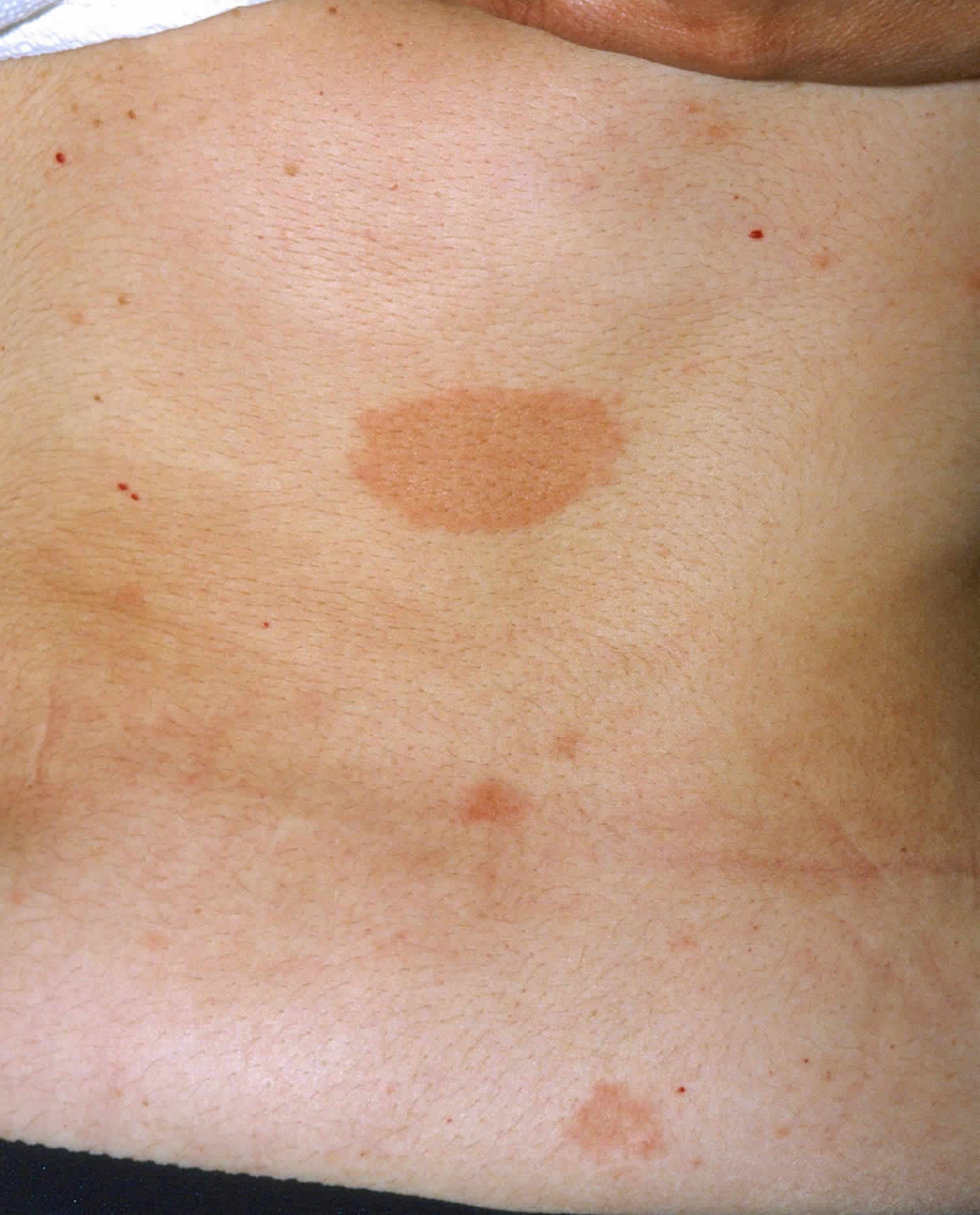
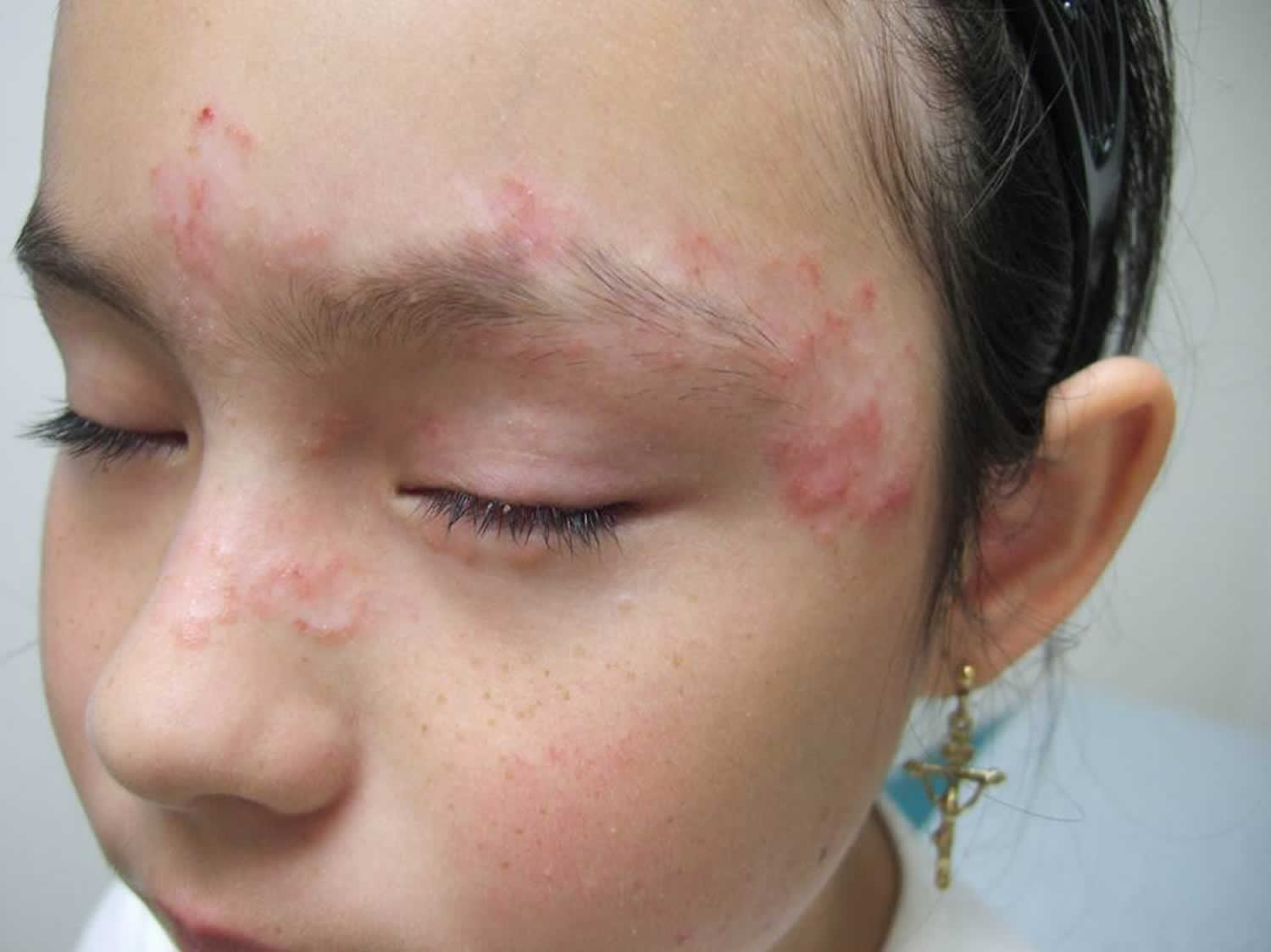
Figure 2. Malar rash in children
What is lupus?
Lupus also known as systemic lupus erythematosus (SLE), like all autoimmune diseases, lupus causes the immune system — your natural protection against foreign invaders like viruses and bacteria — to mistakenly attack your body. What makes lupus unusual, and frequently distressing for patients and families, is its unpredictability: It can affect almost any part of the body, and often many parts at the same time. There are different types of lupus, but in general the word “lupus” is shorthand for the most dominant form, systemic lupus erythematosus (SLE). Systemic means it can involve many parts of your body. Erythematosus comes from the Greek word for “red,” and describes the lupus characteristic cheek rash. Lupus is Latin for “wolf,” which some believe refers to the way a check rash resembles a wolf bite.
Lupus or SLE key points:
- is a chronic (meaning lifelong) autoimmune disorder with no known cause or cure
- affects about five million people worldwide, most often adolescent girls and younger women (15 to 44)
- can target various parts of the body, including the skin, joints, blood and vital organs like the kidneys, heart, lungs and brain
- tends to alternate between being more active (when symptoms surge, or flare) and less active (when symptoms appear to go away) is less common in children
With lupus, doctors can’t predict what part of the body the immune system will choose to strike, or when. But they can use medications to help prevent or blunt these attacks and to extinguish the harmful inflammation. Back in the 1950s, children with lupus faced only about a 30 to 40 percent chance of survival. Today, however, there are powerful medications that can bring this illness under control — often permanently — in the majority of children and allow them to lead full, relatively normal lives.
What are the other forms of lupus?
There are four different types of lupus:
- Systemic lupus erythematosus
- Discoid lupus. This type causes a severe red rash that worsens in sunlight.
- Subacute cutaneous lupus. Skin rashes develop in areas commonly exposed to the sun.
- Drug-induced lupus (DIL). Certain medications can cause lupus symptoms that go away once the drug is stopped.
- Neonatal lupus. This type affects babies of women who have lupus. Skin symptoms usually go away within weeks to a few months after birth. It may cause heart problems.
People of all ages, races and sexes can get lupus, but 9 out of 10 adults with the disease are women between the ages of 15 and 45. African American women are at the highest risk.
Drug-induced lupus
This lupus-like illness can crop up in people who take certain medications for a long time. DIL is fairly rare in children, and when it occurs the most common culprit is the acne drug minocycline. Drug-induced lupus has some of the same symptoms of lupus—fever, fatigue, joint and muscle stiffness—but doesn’t tend to affect vital organs. Symptoms usually disappear within a few weeks after the “triggering” medication is stopped.
Neonatal lupus
This temporary form of lupus affects a small percentage of infants whose mothers have certain lupus autoantibodies. It begins before birth, when these autoantibodies reach the baby via the placenta, and ends within the first several months of life, as the autoantibodies disappear from the baby’s system. The symptoms—skin rash, low blood cell counts—are likewise temporary. However, neonatal lupus does have the potential to cause permanent damage to the baby’s heart (called heart block). If the heart block is significant, a baby may need a pacemaker.
If your child has — or may have — drug-induced lupus or neonatal lupus, his or her doctor will talk with you in detail about what this diagnosis means, and what the next steps will be.
Lupus nephritis
Lupus nephritis is a type of kidney inflammation often found in patients who suffer from systemic lupus erythematosus (SLE) or lupus. SLE, or lupus, is an autoimmune disease that can affect many different parts of the body, including the kidneys.
Lupus nephritis can affect anybody, but it is most common in non-Caucasian females. Depending on age, lupus nephritis can be between 3 to 15 times more common in females than in males, and it is 2 to 3 times more common in women of color.
Lupus nephritis is brought on by lupus or systemic lupus erythematosus, which causes the child’s immune system to attack their own tissues and organs. When the immune system attacks the kidney tissue, it causes inflammation and can destroy parts of kidney.
Types of lupus nephritis
There are six different classifications of lupus nephritis (called Class I through Class VI), sometimes referred to as lupus nephritis stages.
These lupus classifications are based on the form of abnormal cellular structure caused by gene mutation.
What are the signs and symptoms of lupus nephritis?
Lupus nephritis is typically found during the assessment of patients with other signs and symptoms of systemic lupus erythematosus (SLE). Lupus nephritis can also be the first sign that a person has SLE.
Signs and symptoms of systemic lupus erythematosus include:
- Weakness and fatigue
- Malaise (a general feeling of discomfort or illness)
- Fever
- Weight loss
- Joint pain
- Muscle pain
- Butterfly rash (a red rash across the cheeks and bridge of the nose)
- Calcinosis (calcium deposits under the skin)
- Vasculitis (damaged blood vessels)
- Alopecia (hair loss)
- Ulcerations (open sores) in the mouth, nose and sometimes the genitals
Lupus nephritis symptoms are the result of kidney dysfunction. Signs and symptoms of lupus nephritis include:
- Blood in the urine
- Abdominal swelling due to built-up fluid
- Low urine output
- High blood pressure
If your child displays these signs and has not yet been diagnosed with SLE, the next step is to order a laboratory evaluation to tell whether systemic lupus erythematosus is the cause of the kidney dysfunction.
What tests are used to diagnose lupus nephritis?
Your child’s initial laboratory evaluation will include blood and urine tests to assess kidney function.
The tests to diagnose systemic lupus erythematosus (SLE) measure specific immune activity, such as the number and type of antibodies (immune proteins) that are associated with SLE. Your child’s doctor will also look for abnormalities in other immune substances called complement C3 and C4.
If your child is diagnosed with SLE and symptoms suggest that it is affecting the kidney, your child’s doctor will most likely require a kidney biopsy to confirm the lupus nephritis diagnosis.
How is lupus nephritis treated?
Lupus nephritis, as well as systemic lupus erythematosus (SLE) in general, are treated with medications that suppress the immune system. These medications usually include steroids and additional drugs we prescribe very precise dosages and manage closely. Specific treatment regimens depend on the class of lupus nephritis found through the biopsy and the overall extent of how the SLE is affecting the body, outside of the kidneys.
Pediatric lupus causes
Scientists don’t yet know why some children develop lupus and others don’t. Researchers think that people with certain genes are triggered by an external factor, such as stress, a viral infection, medication or regular exposure to chemicals such as silica or pesticides. Since lupus often strikes women during their childbearing years, hormones are believed to play a role.
Lupus not contagious, you can’t “catch” it from another person. Systemic lupus erythematosus is not a disease that parents pass directly down to their children; in fact, there’s only about a 5 percent chance that a son or daughter of someone with lupus will also develop it.
While researchers do believe that genes play a big role in causing lupus, there’s more to it than that. Otherwise, you’d expect that if one identical twin has lupus, the other would, too — but that’s often not the case. Instead, there’s likely a two-part process involved in causing lupus:
- Family history: A child is born with certain genes that make him or her susceptible to lupus. Think of a forest in dry, hot weather: The ingredients for a wildfire are there, but it takes something else to spark the blaze.
- Environmental factors: The child encounters something—or a combination of things—that causes the disease to “ignite.” The environmental factors that may trigger lupus include infections, ultraviolet light and perhaps extreme stress. And given that so many lupus patients are female, it’s also likely that hormones play an important role in the development of and risk for this disease. However, there’s still a lot we don’t know about these triggers, especially why some affect certain children and not others.
Scientists are now working to discover which genes are involved in lupus and how its potential disease triggers work — in order to bring us closer to curing or even preventing this chronic illness.
Pediatric lupus symptoms
Lupus is known as “the great imitator” because many of its earliest warning signs are common in other illnesses, too. Fever, low energy, no appetite? It could be the beginning of lupus — or it might just be the flu.
Lupus is also a very shifty disease. Symptoms often come and go, new ones may crop up, while others seem to disappear. Symptoms also vary greatly from person to person, depending on what part of the body the disease is affecting at the time.
For all these reasons, diagnosing childhood lupus often requires the expertise of pediatric rheumatologists. These specialists are the best qualified to sort out the signs and symptoms of lupus from other diseases, so your child’s treatment can begin as quickly as possible.
Common symptoms of lupus include:
- fatigue
- loss of appetite
- weight loss
- swollen or achy joints
- muscle aches
- fever of over 100 °F (37.8 °C)
- skin rashes, especially a butterfly-shaped rash across the cheeks (this so-called malar rash is a hallmark of lupus) and rashes that develop on sun-exposed skin
- brittle hair, or unusual hair loss
- ulcers in the mouth or nose
- fingers that turn white and/or blue from cold or stress (Raynaud’s phenomenon)
Compared with adults, children with lupus are more likely to have problems with vital organs, especially the kidneys and the brain. These symptoms may include:
- dark urine; swelling around the feet, legs and eyelids and high blood pressure (kidney inflammation, or lupus nephritis)
- shortness of breath, chest pain (lung inflammation, or pleuritis)
- headaches, memory problems, seizures (brain inflammation, or cerebritis)
If your child has symptoms such as fever, fatigue, joint stiffness and skin rashes — especially a butterfly-shaped rash across her cheeks and nose — it might mean she has lupus. You should make an appointment with your child’s pediatrician, who will then make a referral to a rheumatologist if lupus is suspected.
If your child has already been diagnosed with lupus, you should call her specialist about any sudden changes in her symptoms or the appearance of new ones. And remember that infections can potentially be more serious in children with lupus: If your child develops a fever or feels increasingly unwell, let her doctor know right away.
Pediatric lupus diagnosis
You may have read that lupus is extremely difficult to diagnose, and that some patients go a long time, even years, before they know what’s wrong with them. And it’s true the symptoms of lupus can mimic many other illnesses, such as infection and cancer. But if you bring your child to a pediatric rheumatologist — the kind of doctor who knows best what this disease looks like in children — odds are that he or she can determine whether it’s lupus relatively quickly, and if treatment is needed, it can begin right away.
Since there is no single symptom or test result that points to lupus, your child’s doctor will collect a lot of information to make a diagnosis. He or she will conduct a thorough physical exam, make a list of your child’s current symptoms and talk with you about your child’s medical history and the medical history of close family members.
Your child’s doctor will also use certain lab tests to help make a diagnosis and, later, to keep tabs on how the lupus is progressing. These tests include:
- complete blood count (CBC), which is a collection of tests measuring the size, number and maturity of different blood cells in a specific amount of blood. Two important tests include:
- white blood cell count (WBC): This tests indicates the number of white blood cells present. Low levels may point to an active problem with the immune system, like lupus. High levels, on the other hand, may indicate an infection.
- hematocrit (Hct): Indicates the number of red blood cells present. Anemia, or low levels of red blood cells, is often a symptom of lupus.
- antinuclear antibody(ANA), which detects certain abnormal proteins, called antinuclear antibodies, that the immune system often makes when attacking the body’s own tissues. The presence of these antibodies is common in lupus and other autoimmune diseases. However, testing positive for ANA does not equal lupus. Positive tests are also seen in children with other conditions, and even in a handful of perfectly healthy children.
- anti-DNA, which detects a specific antinuclear autoantibody commonly seen with lupus nephritis.
- complement (C3 and C4), which measure blood complement levels. The complement system includes a group of proteins that are part of the immune system. Low levels of complement may indicate lupus activity, and increase the risk for infection.
- C-reactive protein (CRP), which measures the amount of a protein made in the liver. CRP levels tend to increase when there’s an inflammatory process. CRP levels rise very quickly, and may indicate lupus activity or may reflect a new infectious process somewhere in the body.
- erythrocyte sedimentation rate (ESR or sed rate), which measures how quickly red blood cells fall to the bottom of a test tube. If the cells to clump together and fall more rapidly than normal, it can signal there is inflammation in the body.
Your child’s doctor may order other lab tests or imaging tests to check for signs of lupus in specific organs. A urinalysis, for example, can help show whether lupus is affecting the kidneys, while a chest x-ray may show telltale inflammation around the heart or lungs.
Sometimes a biopsy can be helpful in making a diagnosis or evaluating the health of a specific organ or tissue. Almost any part of the body can be biopsied—in which a small sliver of tissue is removed and examined under a microscope— although in lupus the sample is taken from a rash or the kidneys when symptoms are active.
The 11 criteria for lupus
Since lupus symptoms vary so widely and test results don’t always tell the full story, you may wonder how doctors are able to put the puzzle pieces together to come up with a diagnosis. Much of it depends on their past experience with patients, but they also bear in mind 11 lupus criteria laid out by the American College of Rheumatology.
Typically, at least four of the following things must be present for a doctor to diagnose lupus:
- malar rash: a butterfly-shaped rash across cheeks and nose
- discoid rash: raised, scaly patches on the skin
- photosensitivity: skin rash caused by sun exposure
- oral ulcers: small, usually painless sores in the mouth
- arthritis: swelling and achiness in at least two joints
- cardiopulmonary problems: inflammation around the heart and/or lungs
- neurological problems: such as seizures and/or psychosis
- kidney problems: such as blood in the urine
- hematologic (blood) problems: low levels of red blood cells (anemia), white blood cells or platelets
- positive antinuclear antibody (ANA) test
- other positive blood tests that may indicate an autoimmune disease. Antiphospholipid antibodies, including cardiolipin, are common in lupus.
It’s not unusual, though, for experienced physicians to make a diagnosis even when fewer than four criteria are present.
Pediatric lupus treatment
There is no cure for lupus. Treatment involves managing systemic inflammation with medications and lifestyle changes. Treating an unpredictable disease like lupus is like fighting a fire. Doctors can’t know where it might spread, so they focus on what’s actually “on fire”—the places in your child’s body where lupus is active right now. If lupus is affecting your child’s kidneys and central nervous system, for example, the treatment will be very different from what it might be if the disease is affecting your child’s skin. This is why it’s essential to let your child’s doctor know when new symptoms appear, since they could mean another part of the body is under attack.
The medications used to treat lupus fall into two main categories:
- Non-immunosuppressants tend to be milder drugs that fight inflammation or help ease discomfort, and have few side effects.
- Immunosuppressants are much more powerful drugs aimed at bringing the malfunctioning immune system under control. Some have significant side effects and—because they suppress the immune system — all increase the risk of infection.
Other medications may be used to control conditions that are common in people with lupus:
- Water pills that help to prevent fluid buildup.
- Anti-hypertensive drugs to treat high blood pressure.
- Anticonvulsants to prevent or treat seizures.
- Antibiotics for infections.
- Bone-strengthening drugs for osteoporosis.
Nonimmunosuppressants
- Antimalarials or disease-modifying antirheumatic drugs (DMARDs): DMARDs can modify the course of the disease, prevent progression and slow joint damage. DMARDs are often used with nonsteroidal anti-inflammatory drugs (NSAIDs). Most commonly prescribed DMARD is hydroxychloroquine (Plaquenil) which helps reduce the frequency of lupus flares. They are among the safest, most gentle drugs for lupus, yet go a long way toward preventing its life-threatening complications. Most children can expect to be on antimalarials for an indefinite period of time. A potential side effect of hydroxychloroquine is eye problems, but this can be monitored through twice-yearly visits to an ophthalmologist.
- Nonsteroidal anti-inflammatory drugs (NSAIDs) ease symptoms like pain, swelling and stiffness, and are used mainly for kids with lupus arthritis. Among the most common NSAIDs are ibuprofen and naproxen, given in therapeutic doses (that is, higher than the over-the-counter versions). Potential side effects, like stomach problems, are monitored for, but tend to be mild.
Immunosuppressants
- Corticosteroids. Corticosteroids are powerful anti-inflammatory drugs. These drugs are given at the lowest possible dose for the shortest length of time because of side effects. Corticosteroids most often prednisone come in pill form or may be given as an injection into the joint or muscle. Corticosteroids aren’t the same as the anabolic steroids that athletes sometimes take. These are powerful, fast-acting drugs that suppress the entire immune system. Many kids with lupus will need corticosteroids at some point. However, doctors work to phase them out as soon as possible because of their potential side effects, which can include weight gain, facial puffiness (a “cushingoid” appearance), acne, high blood pressure and reduced bone density.
- Steroid-sparing therapies offer many of the benefits of corticosteroids, usually with fewer side effects, but may take much longer to work. You may also hear these drugs called DMARDs, for disease-modifying anti-rheumatic drugs. This group includes methotrexate;azathioprine (brand name Imuran); and mycophenolate mofetil, or MMF (CellCept). Side effects vary by medication, but may include liver problems and anemia.
- Biologics are a relatively new class of steroid-sparing therapies that are based on compounds made by living cells. Instead of suppressing the entire immune system, biologics are more like smart bombs—they only target certain parts of it. Biologics used in lupus include rituximab (Rituxan), tocilizumab (Actemra) and belimumab (Benlysta). Side effects vary by medication.
- Cytotoxins are drugs that destroy rapidly dividing cells in the overactive immune system. Cytotoxins — such as cyclophosphamide (Cytoxan)—are so potent they’re usually reserved for lupus patients with serious kidney disease or central nervous system problems. Side effects may include nausea and vomiting, hair loss and bladder problems.
Aside from medications, your child’s doctor may also prescribe IVIg (intravenous immunoglobulin), which is a blood product made up of healthy antibodies that is delivered by IV, and can help get the immune system back on track. Much more rarely, your child may need to undergo plasmapheresis, a process that removes autoantibodies from the blood. But because it also takes away normal antibodies, it significantly weakens the immune system. That’s why doctors treat only very severe lupus with plasmapheresis.
Complications of lupus require their own medications and treatment procedures, too—dialysis for serious kidney disease, for instance. Your child’s doctor will discuss these with you in detail if and when any complications arise.
Medicine is essential, but it’s not the sum total of your child’s treatment for lupus. Many kids with lupus also require physical and occupational therapy, to increase their mobility and muscle strength and to learn ways to make day-to-day activities easier on their bodies. And because chronic illnesses like lupus can be mentally and emotionally tough to deal with, psychotherapy or counseling can be valuable in helping kids keep the positive outlook they need to beat their disease.
Alternative therapies
When your child is facing a chronic illness like lupus, it’s understandable that you would want to explore all the treatment options. But despite what you may read on the Internet, or hear from friends of friends, there is no “hidden” cure for lupus. No magic herb or special diet will restore your child to perfect health.
That said, there are some things outside conventional medicine — like acupuncture or meditation — that are fairly well studied and do seem to help some people with lupus.
A few words of caution before embarking on any alternative therapies, however:
- Talk it over with your child’s doctor. It’s important that whatever you have in mind won’t interfere with the overall treatment plan. Also, the doctor may know other patients who have tried the therapy— and whether it helped them.
- Don’t skip prescribed treatments. Alternative therapies are meant to supplement traditional medicine, not replace it. It’s vital that your child stick to her medications, or else risk letting her illness get out of control.
- Avoid fly-by-night practitioners. For things like reiki, acupuncture and massage, always ask your doctor for a referral, if appropriate, or recommendations on how to find a qualified care provider.
- And be prepared to pay: By and large, health insurance plans don’t cover alternative therapies.
Home remedies
Lupus is chronic, incurable and unpredictable. But it’s not unbeatable. And you can fight it by helping your child make some simple lifestyle changes at home.
- Eat a healthful diet. Make sure your child is getting plenty of fruits and vegetables, whole grains, low-fat dairy products and lean sources of protein. And load up on Vitamin D and calcium, especially if your child is taking corticosteroids, which can weaken bones.
- Stay active. Encourage your child to walk, swim, bicycle and simply get out and play. While she’ll have to be careful not to get overtired, exercise helps keep muscles strong and joints flexible. And since all lupus patients are at greater risk for cardiovascular disease later in life, making a habit of exercising now will mean better heart health in adulthood.
- Balance rest and physical activity: When disease is active and joints are painful, swollen or stiff, it is important to rest to reduce inflammation and fatigue. When disease activity is low, regular exercise can ease pain and reduce stress. A balanced program includes low-impact aerobic activity, muscle strengthening and flexibility exercises.
- Beware of sun exposure. Because the ultraviolet (UV) rays in sunlight or or fluorescent lights can trigger a flare, your child should wear plenty of broad-spectrum sunscreen (SPF 30 or higher) when she goes outside. Protective clothing like hats and long-sleeved shirts are also strongly recommended. Avoid being outside at peak UV hours between 10 a.m. and 4 p.m.
- Get plenty of rest. People with lupus are prone to fatigue, so remind your child to take a break if she’s feeling run-down — a brief nap can be tremendously helpful in recharging a kid’s batteries. And a full night’s sleep is essential.
- Pace yourself: Fatigue is a common lupus symptom. It’s important to pace yourself to prevent extreme tiredness from taking over your day. Getting restful sleep will help to keep tiredness at bay.
- Reduce stress: Develop strong positive relationships so you have emotional support for managing the ups and downs of a chronic disease. Use relaxation techniques, such as meditation, deep breathing or yoga to reduce stress. Stay connected to activities you enjoy to promote a positive mood.
- Avoid smoking: Smoking including secondhand smoke harms the body and can worsen lupus symptoms.
By doing these things and becoming a full partner in your child’s health care—keeping clinic appointments, alerting doctors to any new symptoms—you may end up feeling more in control of what can be a very daunting illness. The outlook for children with lupus can vary a great deal, however, depending on when the disease begins (called the onset).
- age 15-18 (more common): In this group, pediatric systemic lupus erythematosus tends to look and behave more like adult lupus; its severity can range from mild symptoms to life-threatening disease.
- younger than 15 ( less common): Symptoms can be more severe in this group, and there’s a greater chance that vital organs will be affected.
- younger than than 5 (rare):The smallest children are typically among the sickest lupus patients, largely because most also have complement deficiency, meaning they don’t have enough of certain blood proteins that play a key role in the immune system.
It’s important to remember that everyone with lupus responds to the disease—and the medications used to treat it—in his own unique way. You can’t predict exactly how lupus will progress in your child. But you can greatly improve your child’s odds for a healthy future by helping her understand and stick with the recommended treatments for her condition.
Pediatric lupus prognosis
The outlook for children with lupus can vary a great deal, however, depending on when the disease begins called the onset:
- age 15-18 (more common): In this group, pediatric lupus tends to look and behave more like adult lupus; its severity can range from mild symptoms to life-threatening disease.
- younger than 15 ( less common): Symptoms can be more severe in this group, and there’s a greater chance that vital organs will be affected.
- younger than than 5 (rare):The smallest children are typically among the sickest lupus patients, largely because most also have complement deficiency, meaning they don’t have enough of certain blood proteins that play a key role in the immune system.
It’s important to remember that everyone with lupus responds to the disease—and the medications used to treat it—in his own unique way. You can’t predict exactly how lupus will progress in your child. But you can greatly improve your child’s odds for a healthy future by helping her understand and stick with the recommended treatments for her condition.
References [ + ]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}