



What is Pierre Robin syndrome
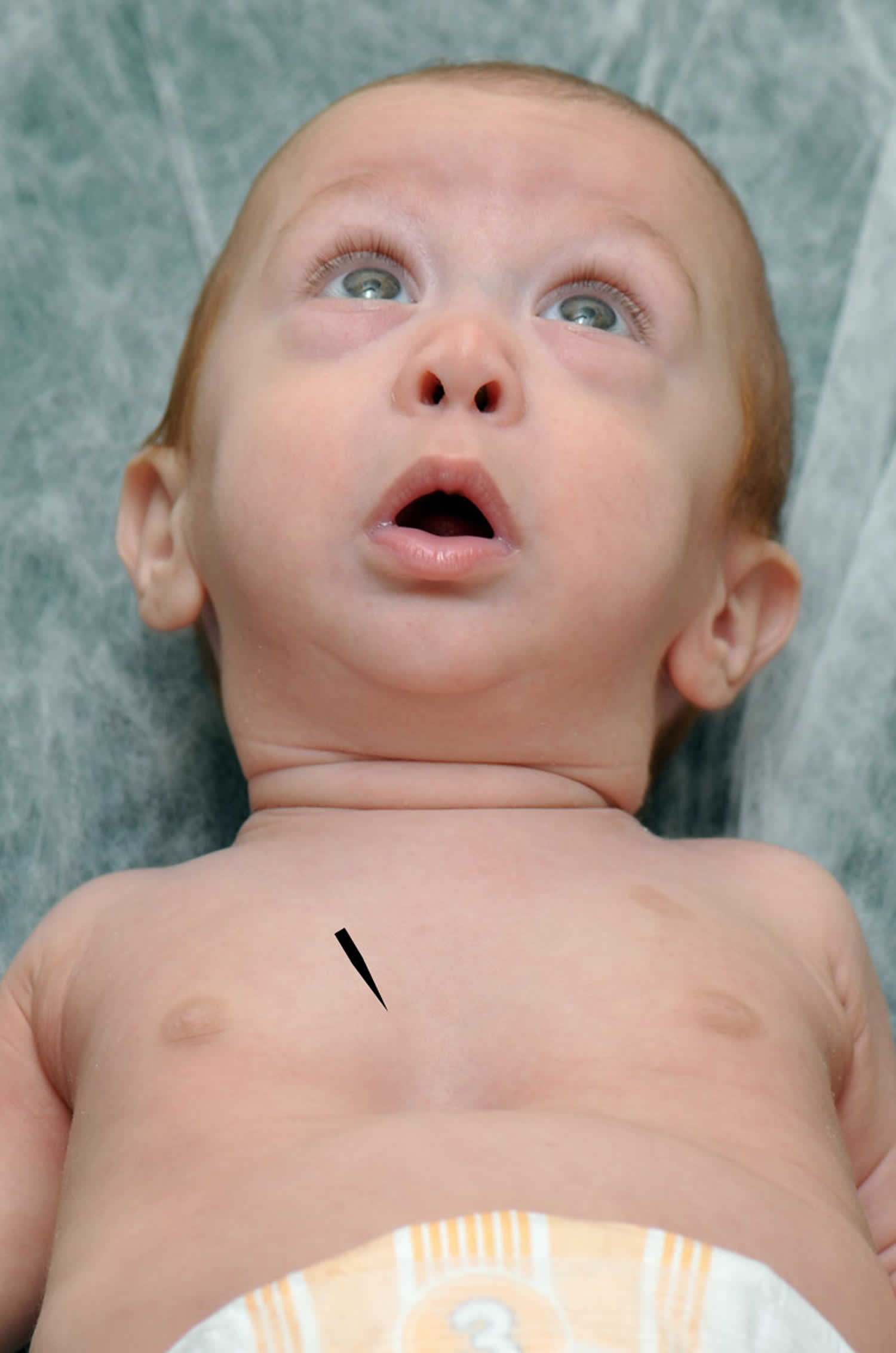
Pierre Robin syndrome is also known as Pierre Robin sequence or Pierre Robin malformation 1). Pierre Robin syndrome is a rare congenital birth defect characterized by a combination of three features: an underdeveloped jaw (micrognathia), backward displacement of the tongue (glossoptosis) and upper airway obstruction. The severity of upper airway obstruction varies, ranging from snoring to life-threatening airway compromise requiring tracheotomy 2). In 80% of the cases of Pierre Robin syndrome, the children also have a U-shaped opening in the roof of their mouths (cleft soft palate). Others may have a high arched palate. While Pierre Robin syndrome is equally common in males and females, there is a higher incidence among twins.
Pierre Robin syndrome was named after Dr. Pierre Robin, a French dental surgeon who first observed its features during the early 20th century.
While the precise cause of Pierre Robin syndrome is not fully clear, the current belief is that multiple contributing factors lead to sequential physical changes within the oral cavity, which ultimately leads to airway obstruction. Pierre Robin syndrome affects approximately 1 per 8,500 to 20,000 neonates 3). The male-to-female ratio is 1:1, except in an X-linked form.
Some people have the features of Pierre Robin sequence as part of a syndrome that affects other organs and tissues in the body, such as Stickler syndrome or campomelic dysplasia. These instances are described as syndromic. When Pierre Robin sequence occurs by itself, it is described as nonsyndromic or isolated. Approximately 20 to 40 percent of cases of Pierre Robin sequence are isolated.
Pierre Robin syndrome is described as a “sequence” because one of its features, underdevelopment of the lower jaw (mandible), sets off a sequence of events before birth that cause the other signs and symptoms. Specifically, having an abnormally small jaw affects placement of the tongue, and the abnormally positioned tongue can block the airways. In addition, underdeveloped jaw (micrognathia) and backward displacement of the tongue (glossoptosis) affect formation of the palate during development before birth, which often leads to cleft palate.
The combination of features characteristic of Pierre Robin syndrome can lead to difficulty breathing and problems eating early in life. As a result, some affected babies have an inability to grow and gain weight at the expected rate (failure to thrive). In some children with Pierre Robin sequence, growth of the mandible catches up, and as adults these individuals have normal-sized chins.
Treatment of Pierre Robin syndrome is multifaceted and individualized, with surgery being performed only to solve the functional problems that a patient may have. Surgical treatments may be indicated for Pierre Robin syndrome patients with more severe clinical conditions, often those associated with airway impairment.
Infants must be kept face down (prone), which allows gravity to pull the tongue forward and keep the airway open. These problems abate over the first few years as the lower jaw grows and assumes a more normal size.
In moderate cases, the patient requires placement of a nasopharyngeal airway (a tube placed through the nose and into the airway) to avoid airway blockage.
In severe cases, surgery is indicated for recurrent upper airway obstruction. A tracheostomy (an operative procedure that creates a surgical airway in the cervical trachea) is sometimes required.
Parents and doctors should continue to monitor child development — particularly jaw and tooth development, growth, and speech.
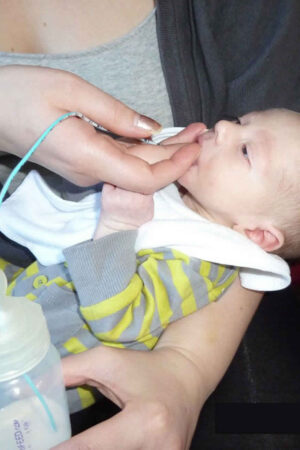
Figure 1. Pierre Robin syndrome (note chest retraction during inhalation due to respiratory distress [arrow])
Why did this happen?
Doctors do no know exactly why Pierre Robin syndrome occurs. They do not believe it is the result of anything the mother did or did not do during pregnancy. If the child only has Pierre Robin syndrome , many experts believe that it is the result of the positioning of the fetus in the early weeks of pregnancy.
Will Pierre Robin syndrome happen to children I have in the future?
Pierre Robin syndrome does not tend to run in families. The chances of you having another child with Pierre Robin syndrome are very small, unless the Pierre Robin Sequence is a part of a syndrome.
What kinds of problems could my child have?
In addition to the physical characteristics common to Pierre Robin syndrome, your child may have the following problems:
- Feeding problems in infancy
- Ear infections
- Reduced hearing
- About 40% of infants with Pierre Robin have Stickler Syndrome and about 15% have Velocardiofacial Syndrome. Doctors recommends genetic testing be done to determine if your infant has either of these associated syndromes. The Cleft Palate Foundation (https://cleftline.org/) has excellent information concerning genetic testing for babies born with Pierre Robin Sequence.
Will my child need surgery?
Depending on the severity of Pierre Robin syndrome, your child may have some or all of the following surgeries:
- Surgery to repair the cleft palate
- Special devices to protect the airway and aid in feeding
- Surgery to improve breathing
- The small jaw associated with Pierre Robin syndrome usually grows out on its own during the first two years, and usually no surgery is necessary on the jaw.
Pierre Robin syndrome life expectancy
Since air and food both pass through the mouth and down the throat, breathing and feeding problems are common. Choking and feeding problems may go away spontaneously over the first few years as the lower jaw grows to a more normal size. There is a significant risk of problems if the airway is not protected against obstruction.
Pierre Robin syndrome possible complications
These complications can occur:
- Breathing difficulties, especially when the child sleeps
- Choking episodes
- Congestive heart failure
- Death
- Feeding difficulties
- Low blood oxygen and brain damage (due to difficulty breathing)
- Type of high blood pressure called pulmonary hypertension
Pierre Robin syndrome causes
Changes in the DNA near the SOX9 gene located in chromosome 17 (17q24) are the most common genetic cause of isolated Pierre Robin syndrome 4). It is likely that changes in other genes, some of which have not been identified, are also involved in Pierre Robin syndrome. Doctors speculate that nongenetic factors, for example conditions during pregnancy that restrict growth of the jaw, may cause some cases of isolated Pierre Robin syndrome. While some studies point to crowding in the uterus or certain neurological conditions.
The SOX9 gene provides instructions for making a protein that plays a critical role in the formation of many different tissues and organs during embryonic development. The SOX9 protein regulates the activity of other genes, especially those that are important for development of the skeleton, including the mandible.
The genetic changes near the SOX9 gene that are associated with isolated Pierre Robin syndrome are thought to disrupt regions of DNA called enhancers, which normally regulate the activity of the SOX9 gene. These changes reduce SOX9 gene activity. As a result, the SOX9 protein cannot properly control the genes essential for normal development of the lower jaw, causing micrognathia (underdeveloped jaw), and consequently, causes displacement of the tongue (glossoptosis), airway obstruction, and, often, formation of a U-shaped cleft palate.
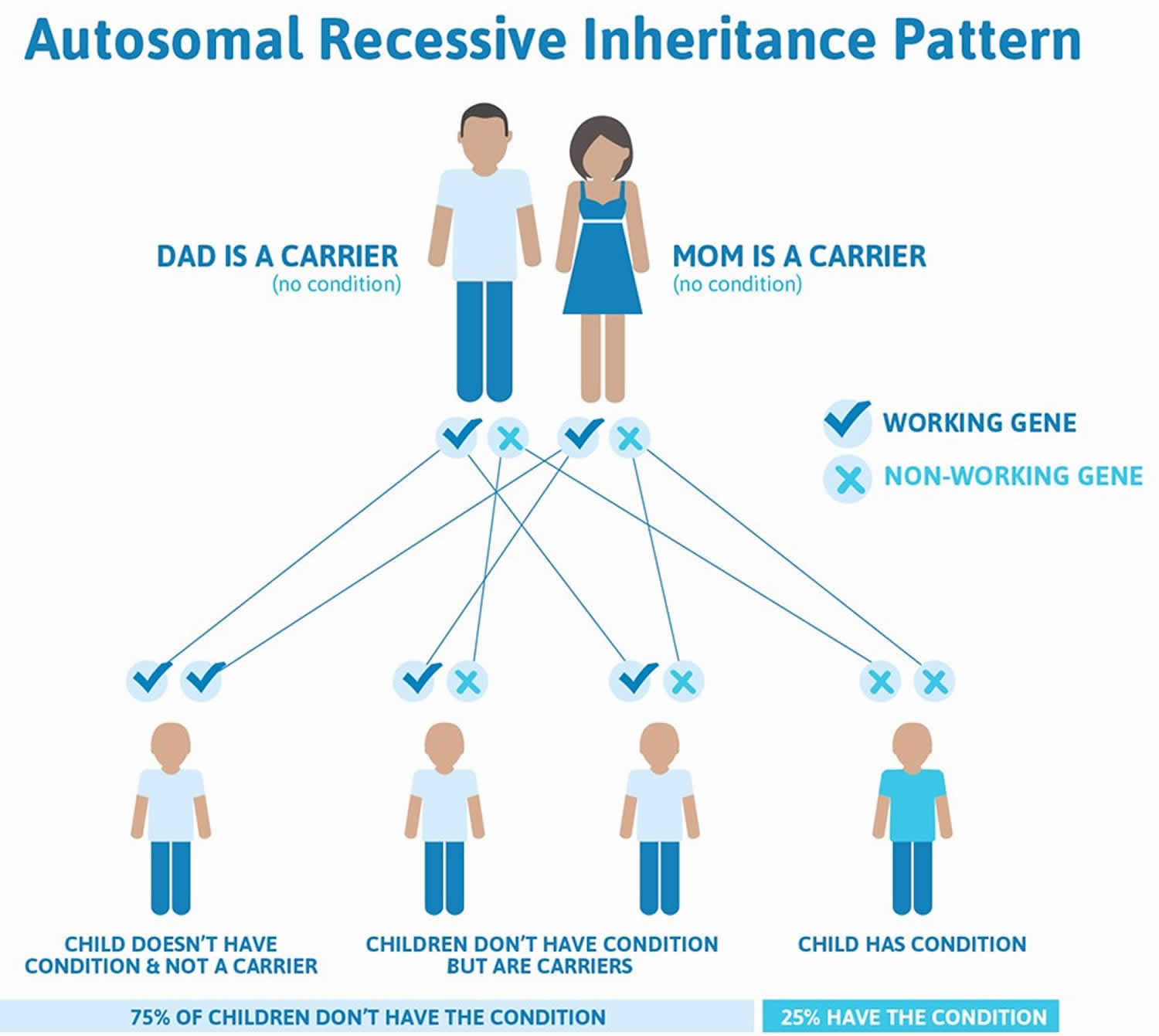
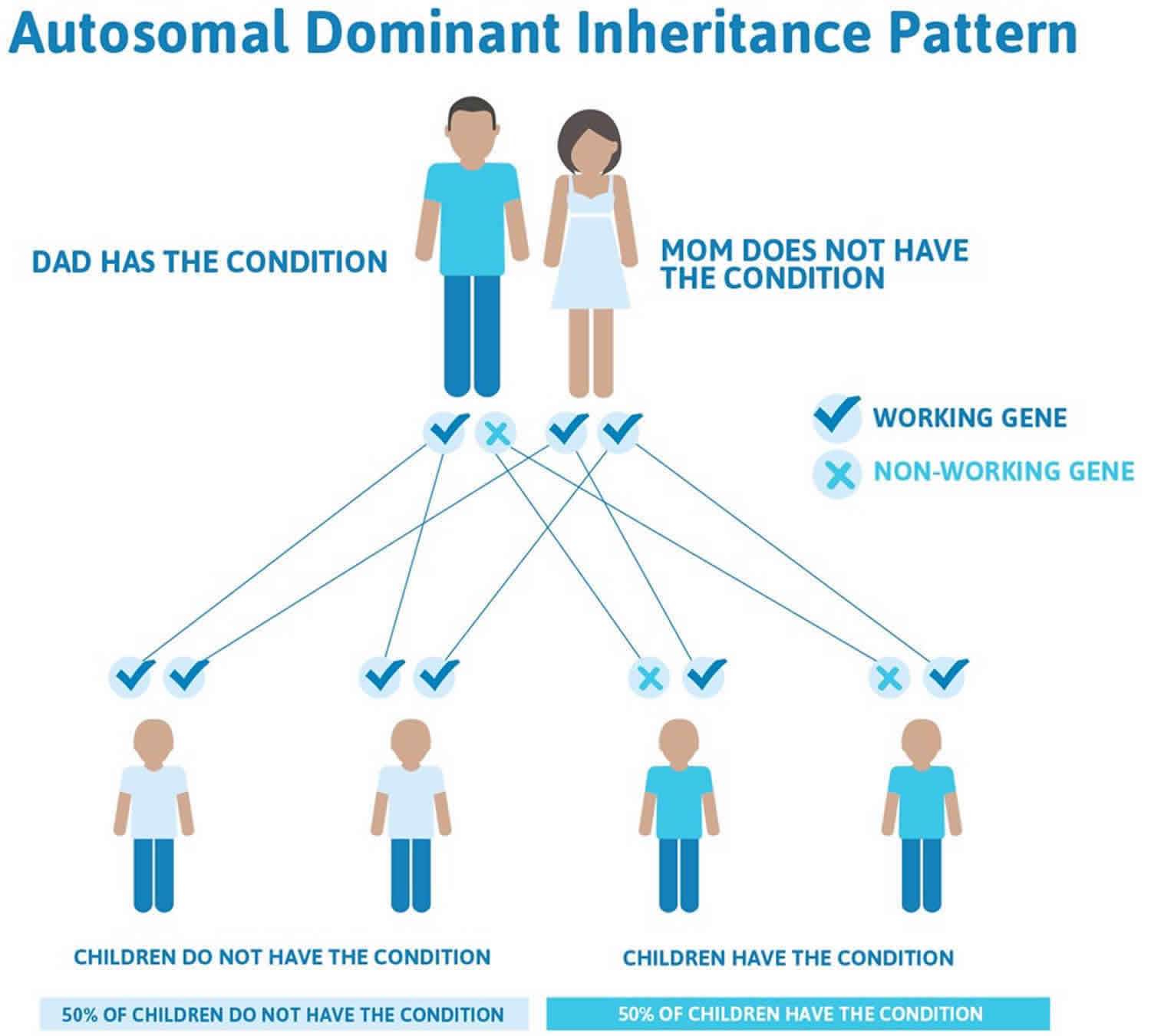
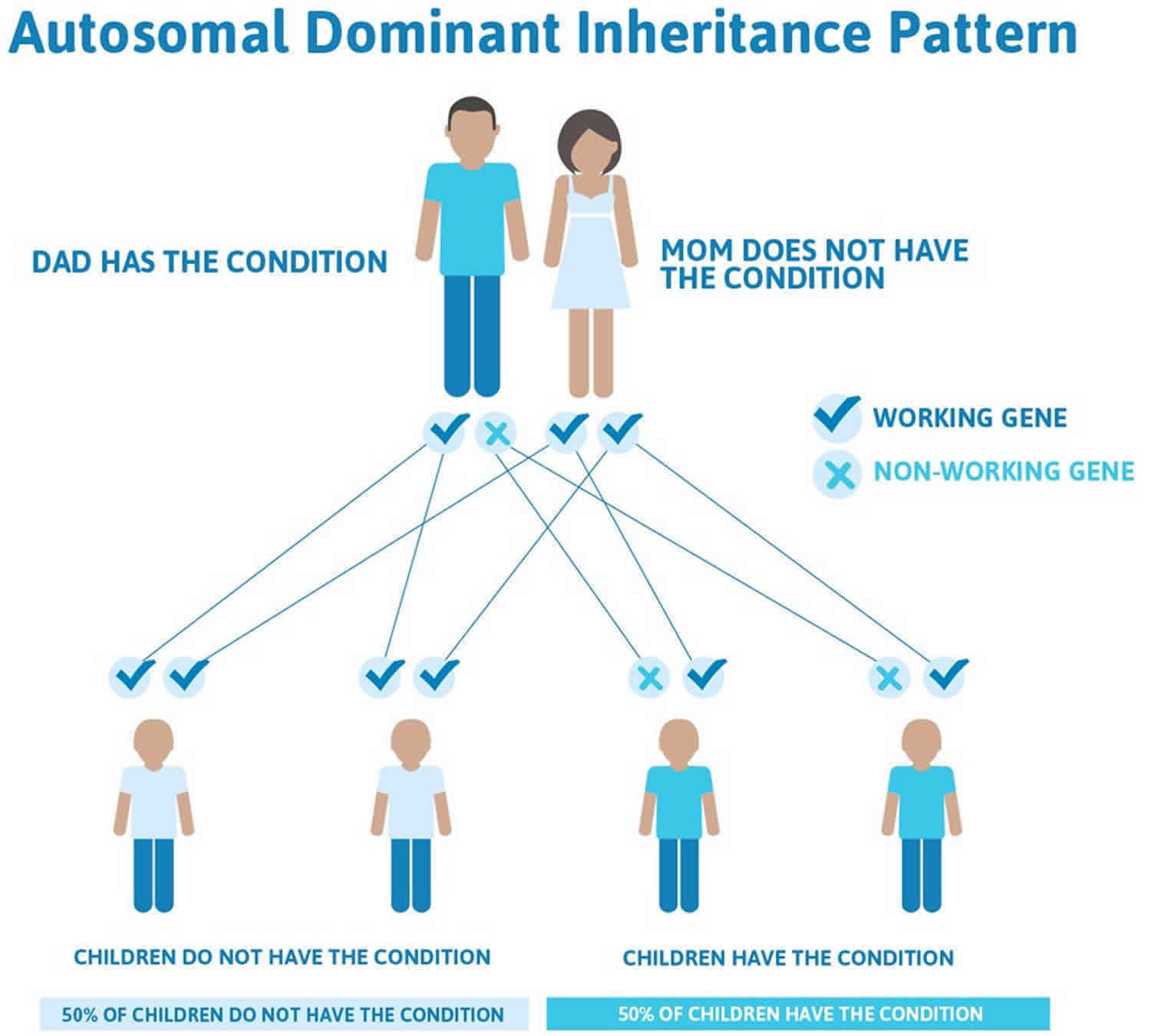
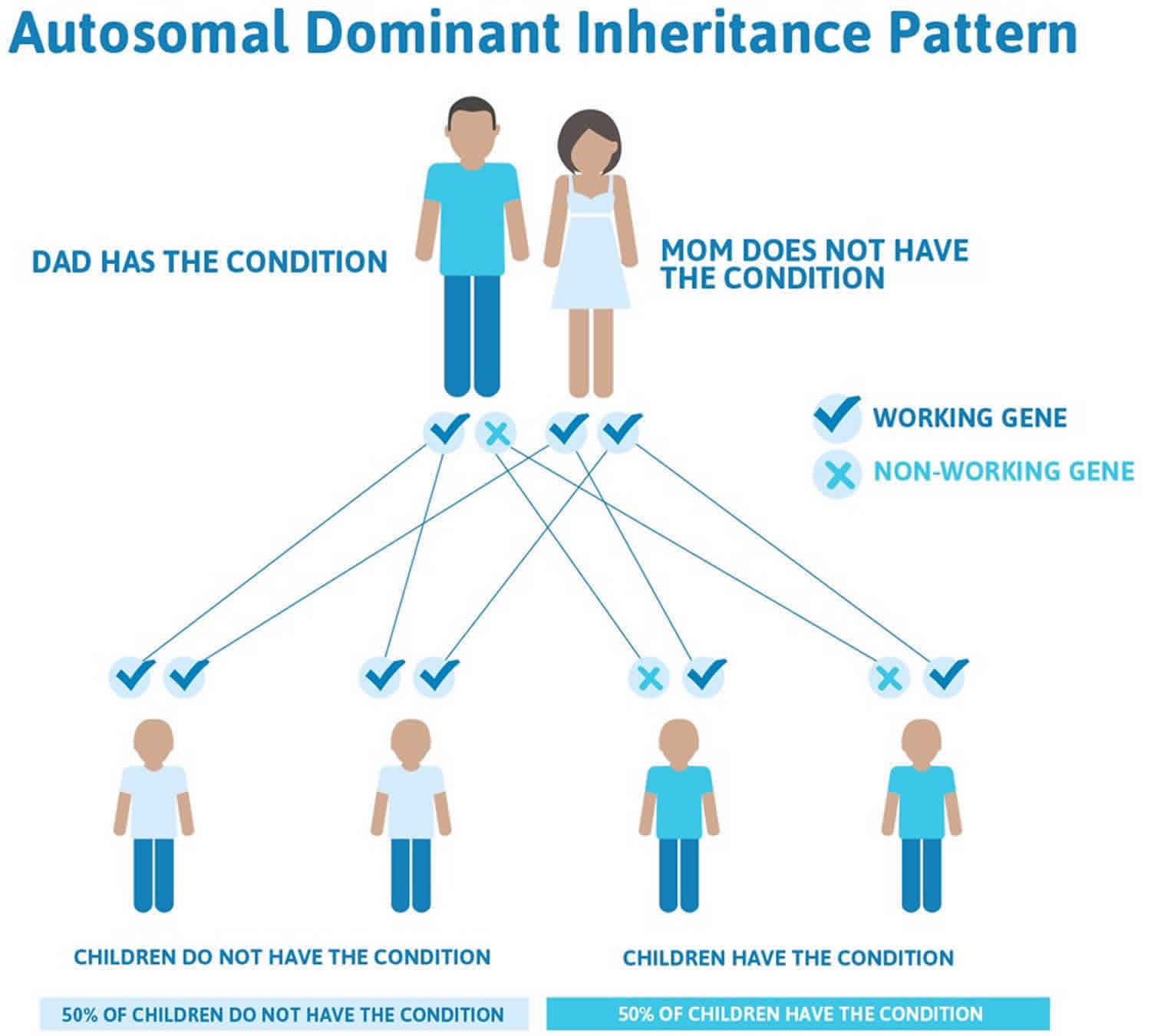
Isolated Pierre Robin sequence is usually not inherited. It typically results from new (de novo) genetic changes and occurs in people with no history of the disorder in their family. When the condition is inherited, it follows an autosomal dominant pattern, which means one copy of the DNA alteration in each cell is sufficient to cause the disorder.
In about 37% of cases, Pierre Robin occurs as part of a syndrome with multiple malformations. Pierre Robin sequence has been reported as occurring in association with Stickler syndrome (20%-25% of these cases), campomelic dysplasia, trisomy 11q syndrome, deletion 4q syndrome, CHARGE association, velocardiofacial syndrome, and Treacher-Collins syndrome 5).
Pierre Robin syndrome symptoms
Babies born with Pierre Robin syndrome commonly experience trouble breathing and feeding early on, resulting from the tongue’s position, smaller jaw size and the cleft palate formation.
Symptoms of Pierre Robin syndrome include:
- Cleft palate
- High-arched palate
- Jaw that is very small with a small chin
- Jaw that is far back in the throat
- Repeated ear infections
- Small opening in the roof of the mouth, which may cause choking or liquids coming back out through the nose
- Teeth that appear when the baby is born
- Tongue that is large compared to the jaw
- Repeated ear infections
- Natal teeth, or teeth that are present at birth
In Pierre Robin syndrome, the lower jaw (mandible) characteristically has an altered shape and position. Typically, it has a reduced length and is located toward the back (microretrognathia). In turn, these changes in the mandible can influence the tongue’s positioning toward the back of the mouth (a ‘retruded’ tongue). Anatomic anomalies of Pierre Robin syndrome also frequently include a U-shaped cleft palate, which affects the dynamics of breathing and speech development.
Specifically, the displacement of the tongue toward the back (posterior) of the mouth predisposes it to fall toward the throat. This may obstruct the airway and cause difficulty breathing. This can vary in severity, ranging from mild disturbance to life-threatening respiratory distress. Airway obstruction can also occur during the night, in the case of a related condition called ‘obstructive sleep apnea’. This is a sleep disorder characterized by breathing that temporarily stops and restarts because of periodic blockage of the airways.
Since food traveling toward the gastrointestinal tract also passes through the mouth and throat, feeding difficulties can also arise due to abnormal oral cavity anatomy. Depending on the severity, this can lead to issues like choking (aspiration) or gaining less weight gain than expected (which doctors refer to as ‘failure to thrive’). There is also a higher prevalence of acid (gastroesophageal) reflux in children with Pierre Robin syndrome.
Other possible manifestations of Pierre Robin syndrome include cardiovascular and lung conditions, such as heart murmurs, high blood pressure in the arteries of the lungs (pulmonary hypertension), and narrowing of the opening between the lung artery and the right ventricle of the heart (pulmonary stenosis). Anomalies of the musculoskeletal system, including those in the arms, legs, feet, and vertebral column, are also common. Inflammation of the middle ear (otitis media) usually accompanied by repeat ear infections occurs in about 80% of patients, and eye (ocular) defects are noted in about 10% to 30% of patients. Teeth present at birth (natal teeth) are another frequent finding.
Pierre Robin syndrome diagnosis
A physical examination is usually sufficient for your health care provider to diagnose Pierre Robin syndrome. A genetics consultation can rule out other associated anomalies and syndromes.
Pierre Robin syndrome treatment
A team of specialists will work together to address affected functions, including breathing, hearing, feeding and sleeping. If your child has Pierre Robin syndrome, you can expect treatment to come in stages. Since the condition affects a variety of functions, including hearing, breathing and feeding, several specialists will be involved in your child’s care.
Breathing
Infants with Pierre Robin syndrome should be observed closely for breathing difficulties.
The first priority will be to keep the upper airway open to allow for proper breathing. Laying your child on his or her stomach (prone position) can help prevent the tongue from falling back toward the throat and blocking off the airway. If placing the infant on his or her stomach does not solve the problem, other treatments aimed at keeping the upper airway open may be recommended. These include a ‘nasopharyngeal airway’ or nasal trumpet (a small tube threaded through the nose into the upper airway).
In cases of severe obstruction, your doctor may recommend surgery to enlarge the lower jaw (so that the tongue can come into the mouth) or a tracheotomy to create an opening in the windpipe to assist the infant in breathing.
Surgery to improve the appearance of the jaw is rarely necessary because the small lower jaw seen at birth most often grows to a more normal size by 18 months of age.
Feeding
Jaw size, tongue placement and cleft palate all contribute to difficulties feeding. Infants with minor degrees of Pierre Robin syndrome can learn to feed using specially adapted nipples and bottles.
However, for babies with more severe Pierre Robin syndrome, the risk of inhaling fluid into the lungs is high. A feeding tube may be recommended as a temporary solution to allow for proper weight gain. While feeding difficulties decrease within the first two years, children that may need long-term assistance could require a gastric tube inserted into the abdominal wall.
Cleft Palate and Hearing Problems
The timing of the cleft palate repair varies depending on the child’s individual growth and development. To close the cleft palate, surgery is typically performed between 12 and 18 months of age. Doctors may postpone the corrective surgery, however, to allow the opening in the palate to close on its own as natural growth occurs.
Cleft palate is repaired with a two- to three-hour surgical procedure and requires a one- to two-night hospital stay. During the procedure, tubes may be inserted into the ear to lessen fluid buildup.
Some children may also require speech therapy following cleft palate repair.
Teeth Problems
Since the lower jaw is smaller in children with Pierre Robin syndrome, teeth crowding is frequently a concern. Orthodontists, pediatric dentists and craniofacial surgeons should work together to monitor dental development.
Symptomatic and supportive treatment
Symptomatic and supportive treatment may be provided using a multidisciplinary team approach, in order to best meet the needs of the affected individual. If speech is impaired, the child should participate in speech therapy or be monitored by a speech pathologist. Ear, nose, and throat doctors (otolaryngologists) and audiologists can provide follow-up on ear- and hearing-related issues. Surgically placed drainage tubes may be recommended if ear infections are recurrent. A combination of orthodontists, maxillofacial surgeons, and dentists may work together to monitor the oral cavity, for example by looking to avoid crowding of the teeth and to ensure proper tooth alignment. Ophthalmology may be consulted to monitor for ocular abnormalities. Genetic counseling may be of benefit for patients and their families.
Pierre Robin syndrome prognosis
All neonates with significant Pierre Robin syndrome are at risk for sudden death. The sudden infant death syndrome (SIDS) data show that the risk of SIDS is increased when infants sleep in the prone position. Neonates with Pierre Robin syndrome already have a compromised airway, and they also typically require prone positioning. Accordingly, monitoring of these neonates should be strongly considered.
Infants with Pierre Robin syndrome deserve to be treated with a multidisciplinary approach that involves a knowledgeable and experienced team capable of providing a comprehensive assessment, a realistic plan of treatment, and appropriate follow-up. Engaging the family in the early stages of the evaluation, the ongoing medical investigations, the issues regarding the child’s care, and future planning generally leads to satisfaction, even in the most difficult of medical scenarios.
In a study of 103 patients followed for a median of 8.6 years (range, 0.1-21.9 years), Logjes et al 6) documented a 10% mortality (n = 10) at a median patient age of 0.8 years (range, 0.1-5.9 years). Of the 10 infants who died, nine had syndromic Pierre Robin syndrome; seven of the nine died of respiratory insufficiency due to various causes, and the other two died of arrhythmia due to hypernatremia and of West syndrome with status epilepticus. The infant with nonsyndromic Pierre Robin syndrome died of brain ischemia after mandibular distraction osteogenesis.
References [ + ]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}