
Contents
Aicardi Goutieres syndrome
Aicardi-Goutieres syndrome also called familial infantile encephalopathy with calcification of basal ganglia and chronic cerebrospinal fluid lymphocytosis, is an inherited disorder that mainly affects the brain, the immune system, and the skin 1). Most newborns with Aicardi-Goutieres syndrome do not show any signs or symptoms of the disorder. However, about 20 percent are born with a combination of features that include an enlarged liver and spleen (hepatosplenomegaly), elevated blood levels of liver enzymes, a shortage of blood cells called platelets that are needed for normal blood clotting (thrombocytopenia), and neurological abnormalities. While this combination of signs and symptoms is typically associated with the immune system’s response to a viral infection that is present at birth (congenital), no actual infection is found in these infants. For this reason, Aicardi-Goutieres syndrome is sometimes referred to as a “mimic of congenital infection.” Aicardi-Goutieres syndrome is a rare disorder. Its exact prevalence is unknown.
There are several types of Aicardi-Goutieres syndrome, depending on the gene that causes the condition: TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR and IFIH1, genes 2). Most Aicardi-Goutieres syndrome cases are inherited in an autosomal recessive pattern, although rare autosomal dominant cases have been reported. Treatment of Aicardi-Goutieres syndrome is symptomatic and supportive. This means that you can treat the symptoms, but there is no cure for the disease 3). The prognosis depends mainly on the severity neurologic problems and in the age of onset of these problems 4).
Loss of white matter in the brain (leukodystrophy) and abnormal deposits of calcium (calcification) in the brain leads to an early-onset severe brain dysfunction (encephalopathy) that usually results in severe intellectual and physical disability 5). Additional symptoms may include epilepsy, painful, itchy skin lesion (chilblains), vision problems, and joint and muscle stiffness (spasticity), involuntary muscle twisting and contractions (dystonia), and weak muscle tone (hypotonia) in the torso. Other signs and symptoms may include a very small head (microcephaly), presence of white blood cells and other sign of inflammation in the cerebrospinal fluid, which is the fluid that surrounds the brain and spinal cord (central nervous system). Symptoms usually progress over several months before the disease course stabilizes.
Within the first year of life, most individuals with Aicardi-Goutieres syndrome experience an episode of severe brain dysfunction (encephalopathy), typically lasting for several months. During this encephalopathic phase of the disorder, affected babies are usually extremely irritable and do not feed well. They may develop intermittent fevers in the absence of infection (sterile pyrexias) and may have seizures. They stop developing new skills and begin losing skills they had already acquired (developmental regression). Growth of the brain and skull slows down, resulting in an abnormally small head size (microcephaly). In this phase of the disorder, white blood cells and other immune system molecules associated with inflammation can be detected in the cerebrospinal fluid, which is the fluid that surrounds the brain and spinal cord (central nervous system). These abnormal findings are consistent with inflammation and tissue damage in the central nervous system.
The encephalopathic phase of Aicardi-Goutieres syndrome causes permanent neurological damage that is usually severe. Medical imaging reveals loss of white matter in the brain (leukodystrophy). White matter consists of nerve fibers covered by myelin, which is a substance that protects nerves and insures rapid transmission of nerve impulses. Affected individuals also have abnormal deposits of calcium (calcification) in the brain. As a result of this neurological damage, most people with Aicardi-Goutieres syndrome have profound intellectual disability. They also have muscle stiffness (spasticity); involuntary tensing of various muscles (dystonia), especially those in the arms; and weak muscle tone (hypotonia) in the torso.
Some people with Aicardi-Goutieres syndrome have features characteristic of autoimmune disorders, which occur when the immune system malfunctions and attacks the body’s own systems and organs. Some of these features overlap with those of another disorder called systemic lupus erythematosus (SLE). A feature of SLE that also occurs in about 40 percent of people with Aicardi-Goutieres syndrome is a skin problem called chilblains. Chilblains are painful, itchy skin lesions that are puffy and red, and usually appear on the fingers, toes, and ears. They are caused by inflammation of small blood vessels, and may be brought on or made worse by exposure to cold. Vision problems, joint stiffness, and mouth ulcers are other features that can occur in both disorders.
As a result of the severe neurological problems usually associated with Aicardi-Goutieres syndrome, most people with this disorder do not survive past childhood. However, some affected individuals who develop the condition later or have milder neurological problems live into adulthood.
Figure 1. Aicardi Goutieres syndrome chilblains

Footnote: (A) Blue toes in a child with AicardieGoutières syndrome caused by RNASEH2B mutations. (B) Less severe—and more typical—chilblain-like lesion on the little toe of a child with RNASEH2C-associated disease.
[Source 6) ]Aicardi Goutieres syndrome causes
Mutations in several genes can cause Aicardi-Goutieres syndrome. Several of these genes, the TREX1, RNASEH2A, RNASEH2B, and RNASEH2C genes, provide instructions for making nucleases, which are enzymes that help break down molecules of DNA and its chemical cousin RNA when they are no longer needed. These DNA and RNA molecules or fragments may be generated during the first stage of protein production (transcription), copying (replication) of cells’ genetic material in preparation for cell division, DNA repair, cell death (apoptosis), and other processes. Mutations in any of these genes are believed to result the absence or abnormal functioning of the respective nuclease enzyme. Researchers suggest that absent or impaired enzyme function may result in the accumulation of unneeded DNA and RNA in cells. The unneeded DNA and RNA may be mistaken by cells for the genetic material of viral invaders, triggering immune system reactions in multiple body systems that result in encephalopathy, skin lesions, and other signs and symptoms of Aicardi-Goutieres syndrome.
Mutations in other genes, including the SAMHD1, IFIH1, and ADAR genes, can also cause Aicardi-Goutieres syndrome. These genes provide instructions for making proteins that are involved in the immune system. Mutations in these genes cause inappropriate activation of the body’s immune response, resulting in inflammatory damage to the brain, skin, and other body systems that lead to the characteristic features of Aicardi-Goutieres syndrome.
Aicardi Goutieres syndrome inheritance pattern
Aicardi-Goutieres syndrome can have different inheritance patterns. In most cases caused by mutations in the ADAR, TREX1, RNASEH2A, RNASEH2B, RNASEH2C, and SAMHD1 genes, it is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
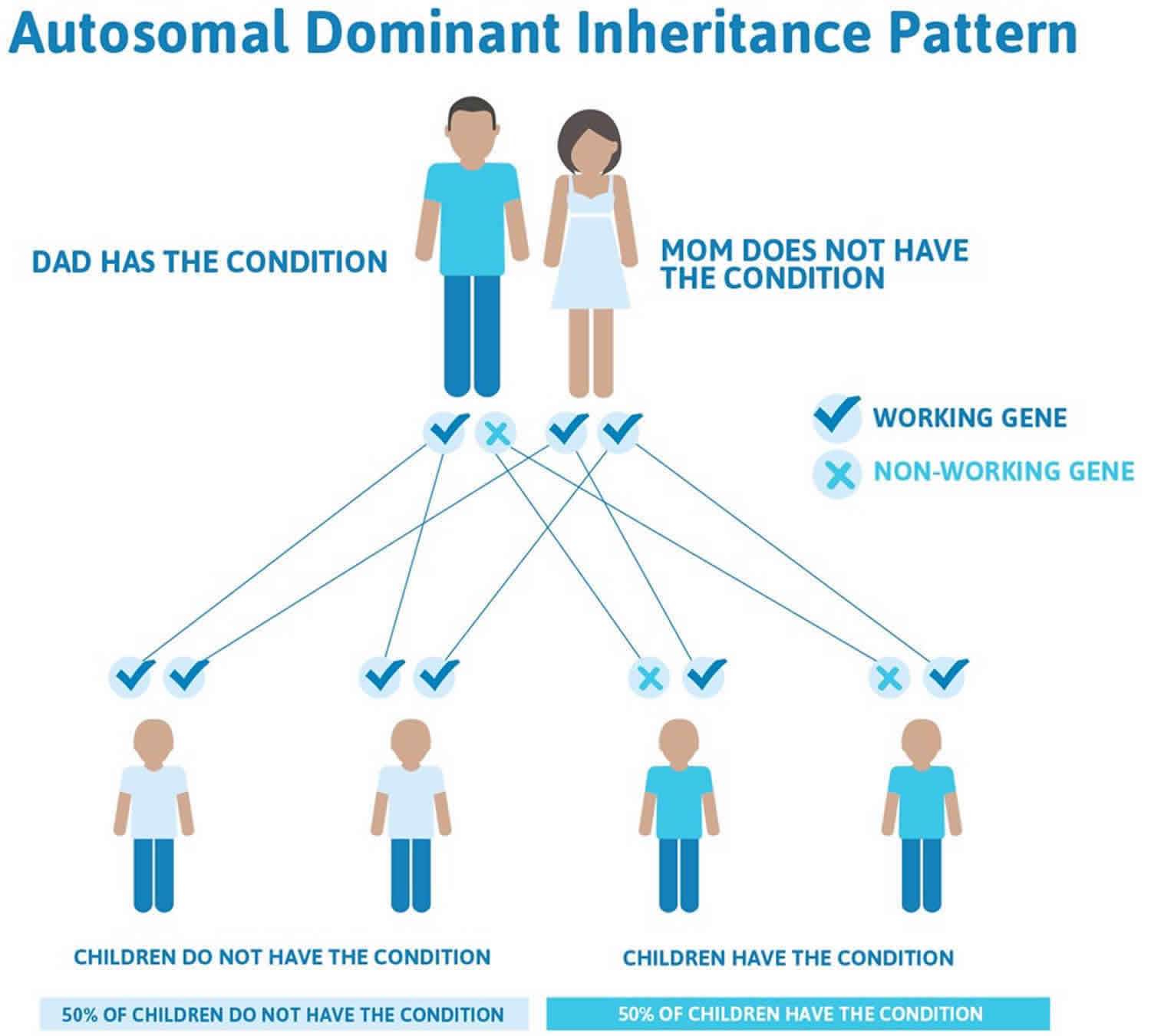
When caused by mutations in the IFIH1 gene or by certain severe mutations in the TREX1 or ADAR gene, Aicardi-Goutieres syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. These cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
Figure 2. Aicardi-Goutieres syndrome autosomal recessive inheritance pattern
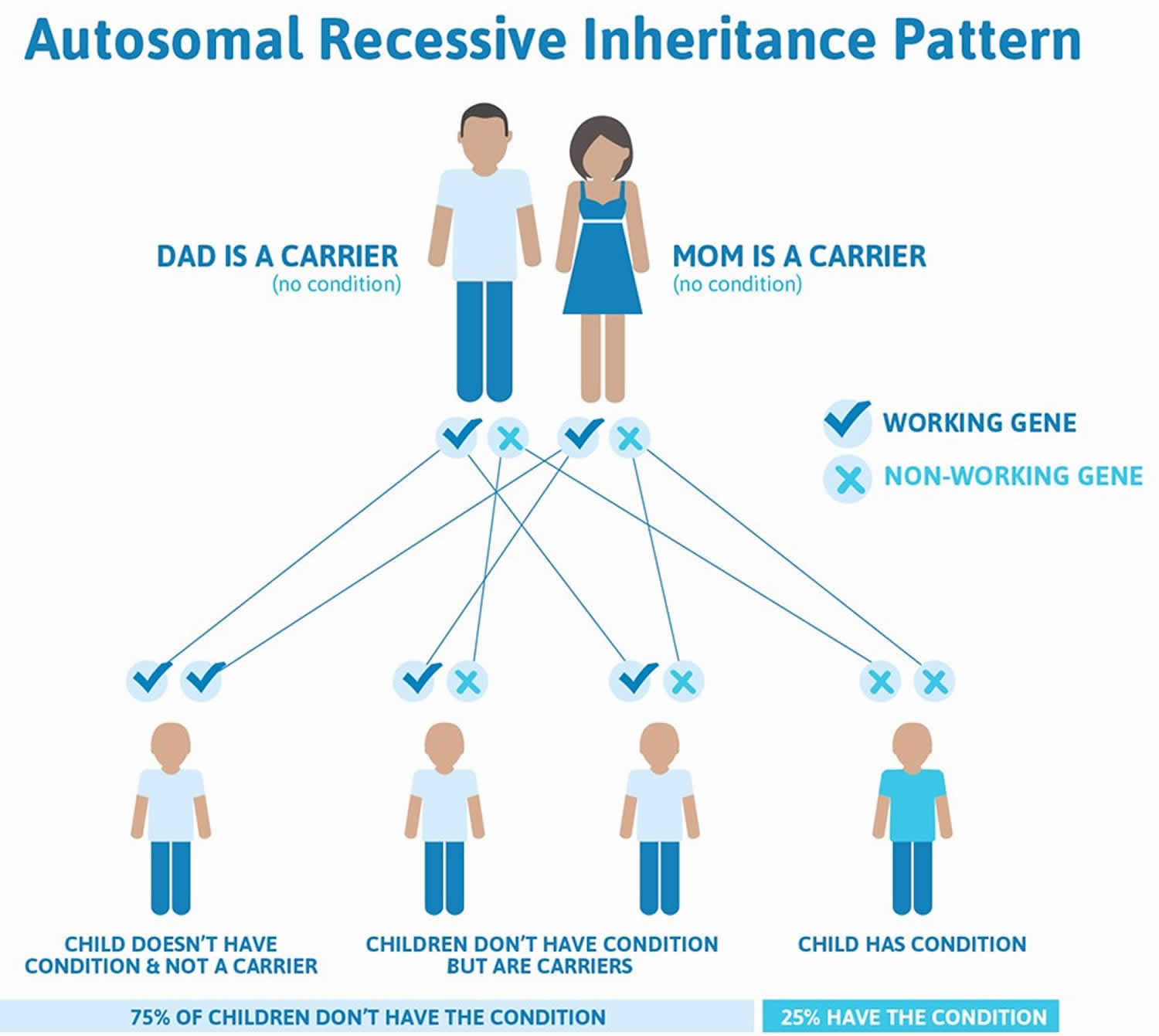
Figure 3. Aicardi-Goutieres syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Aicardi Goutieres syndrome symptoms
In its most characteristic form, Aicardi-Goutieres syndrome can be considered an early-onset encephalopathy associated with significant intellectual and physical disability. Symptoms of Aicardi-Goutieres syndrome usually appear within the first six months of life. Aicardi-Goutieres syndrome is generally either fatal or else it results in a persistent vegetative state in early childhood. Generally, the first symptoms observed are vomiting, feeding difficulties, and lack of progress in motor and social skills. A subset of patients has a later onset of disease, which usually occurs between six-twelve months of age, and is marked by loss of previously acquired motor skills and spasticity. The course of the disease is severe and progressive; death occurs in 25% of patients before 17 years of age. However, in some cases, there can be less impairment, and some retention of contact with surroundings and social interactions. Below is a list of symptoms that may be present for Aicardi-Goutieres syndrome, along with definitions as necessary. Please note that all of these symptoms are not present in all cases.
- Microcephaly: abnormally small head
- Early progressive encephalopathy: abnormalities of the brain
- Lack of progress of motor and social skills; no or very poor contact with surroundings
- Feeding difficulties
- Irritability
- Vomiting
- Spasticity: presence of spasms
- Dystonia: Abnormal muscle tone, characterized by prolonged, repetitive muscle contractions that may cause twisting or jerking movements of the body or a body part.
- Visual Inattention
- Ocular jerks: abnormal eye movements
- Sterile CSF lymphocytosis: cerebrospinal fluid that has elevated levels of lymphocytes (a certain cell of the immune system), but in which there are no indications of infection (sterility)
- Skin lesions of the toes, fingers, ear lobes looking like chilblains (itchy red swelling of the skin), puffy hands and feet, and cold feet
- Intracerebral calcification: presence of calcium deposits on a particular area of the brain.
Pregnancy, delivery, and the neonatal period are normal in approximately 80% of infants with Aicardi-Goutieres syndrome 7). However, brain calcifications can be identified in utero 8) and 20% of cases, mainly those caused by biallelic pathogenic variants in TREX1, present at birth with abnormal neurologic findings, hepatosplenomegaly, elevated liver enzymes, and thrombocytopenia, a picture reminiscent of congenital infection.
All other affected infants present at variable times after the first few weeks of life, frequently after a period of apparently normal development. The majority of these later-presenting infants exhibit subacute onset of a severe encephalopathy characterized by extreme irritability, intermittent sterile pyrexias, loss of skills, and slowing of head growth. The encephalopathic phase usually lasts several months. The opinion of most pediatricians caring for such children is that the disease does not progress beyond the encephalopathic period; occasionally, however, affected individuals do appear to show progression and/or episodes of regression. Death is usually considered to be secondary to the neurologic damage incurred during the initial disease episode, not to further regression. Several affected individuals older than age 30 years show no obvious signs of disease progression.
Neurologic features. Typically, affected individuals have peripheral spasticity, dystonic posturing (particularly of the upper limbs), truncal hypotonia, and poor head control. Seizures are reported in up to half of affected children, but are usually relatively easily controlled 9). A number of children demonstrate a marked startle reaction to sudden noise, and the differentiation from epilepsy can be difficult. Most affected individuals have severe intellectual and physical impairment. Variability in the severity of the neurologic outcome can be observed among sibs. Most affected children exhibit a severe acquired microcephaly; in children with preserved intellect head circumference can be normal.
Hearing is almost always normal.
Ophthalmology. Visual function varies from normal to cortical blindness. Ocular structures are almost invariably normal on examination. However, there is a risk of congenital glaucoma or later-onset glaucoma 10).
Skin lesions. As many as 40% of affected individuals 11) have skin lesions with chilblains on the fingers and toes and sometimes the ears and other pressure points (e.g., elbows) 12). The cutaneous lesions may be complicated by periungual infection and necrosis.
Other
- Intracranial large-vessel disease. An additional previously undescribed feature of Aicardi-Goutieres syndrome, which so far appears to be almost exclusively related to biallelic pathogenic variants in SAMHD1, is intracranial large-vessel disease causing both intracranial stenoses (in some cases reminiscent of moyamoya syndrome) and aneurysms 13).
- Refractory four-limb dystonia. Several individuals with ADAR pathogenic variants have been reported to demonstrate an acute or subacute onset of refractory four-limb dystonia starting between age eight months and five years 14).
- Individuals can be developmentally normal at initial presentation.
- This phenotype can occur due to either biallelic pathogenic variants in ADAR or the autosomal dominant pathogenic variant p.Gly1007Arg in ADAR.
- Like other Aicardi-Goutieres syndrome-related phenotypes, bilateral striatal necrosis due to biallelic pathogenic variants in ADAR or the recurrent dominant pathogenic p.Gly1007Arg variant in ADAR1 are typically associated with an upregulation of ISGs.
Infrequent features seen in a cohort of 123 individuals with molecularly confirmed Aicardi-Goutieres syndrome
- Scoliosis
- Cardiomegaly
- Abnormal antibody profile
- Preserved language
- Demyelinating peripheral neuropathy
- Congenital glaucoma
- Micropenis
- Hypothyroidism
- Insulin-dependent diabetes mellitus
- Transitory deficiency of antidiuretic hormone
Aicardi Goutieres syndrome diagnosis
Aicardi-Goutieres syndrome is difficult to diagnose, as many of the symptoms overlap with other disorders. In its most characteristic form, Aicardi-Goutières syndrome can be considered an early-onset encephalopathy associated with significant intellectual and physical disability. The clinical symptoms of the disease are taken into consideration, as well as certain brain abnormalities seen by MRI. A sample of the cerebrospinal fluid (CSF) will be taken from a spinal tap. This fluid can then be tested for increased levels of a certain type of cell of the immune system (lymphocytes), a condition known as chronic lymphocytosis. These cells are normally only elevated during infection, so the combination of lymphocytosis combined with a lack of evidence of infection can support a diagnosis of Aicardi-Goutieres syndrome. The CSF may also be tested for elevated levels of a molecule known as interferon-gamma, which can also be suggestive of this disease.
Suggestive findings
Aicardi-Goutieres syndrome should be suspected in individuals with the following clinical, neuroimaging, and supportive laboratory findings 15).
Clinical features
- Encephalopathy and/or significant intellectual disability
- Acquired microcephaly during the first year of life
- Dystonia and spasticity
- Sterile pyrexias
- Hepatosplenomegaly
- Chilblain lesions on the feet, hands, ears, and sometimes more generalized mottling of the skin.
Exclusion criteria include the following:
- Evidence of prenatal/perinatal infection including, but not limited to, CMV, toxoplasmosis, rubella, herpes simplex, Zika, and HIV
- Evidence of a known other metabolic disorder or neurodegenerative disorder
Neuroimaging
- Calcification (best visualized on CT scan) of the basal ganglia, particularly the putamen, globus pallidus and thalamus but also extending into the white matter, sometimes in a para- (rather than true peri-) ventricular distribution 16). See Figure 4.
- Note: Intracranial calcification is not always recognized on MRI, the initial imaging modality employed in most units.
- White matter changes, particularly affecting the frontotemporal regions with (in severe cases) temporal lobe cyst-like formation. See Figure 5. On MRI, appears on T2-weighted images as a hyperintense signal most commonly located around the horns of the ventricles.
- Cerebral atrophy, which may be progressive and involve the periventricular white matter and sulci. Cerebellar atrophy and brain stem atrophy may also be prominen 17).
- Bilateral striatal necrosis
- Intracerebral vasculopathy including intracranial stenosis, moyamoya, and aneurysms
Figure 4. Cerebral calcifications on CT scan in individuals with Aicardi-Goutieres syndrome
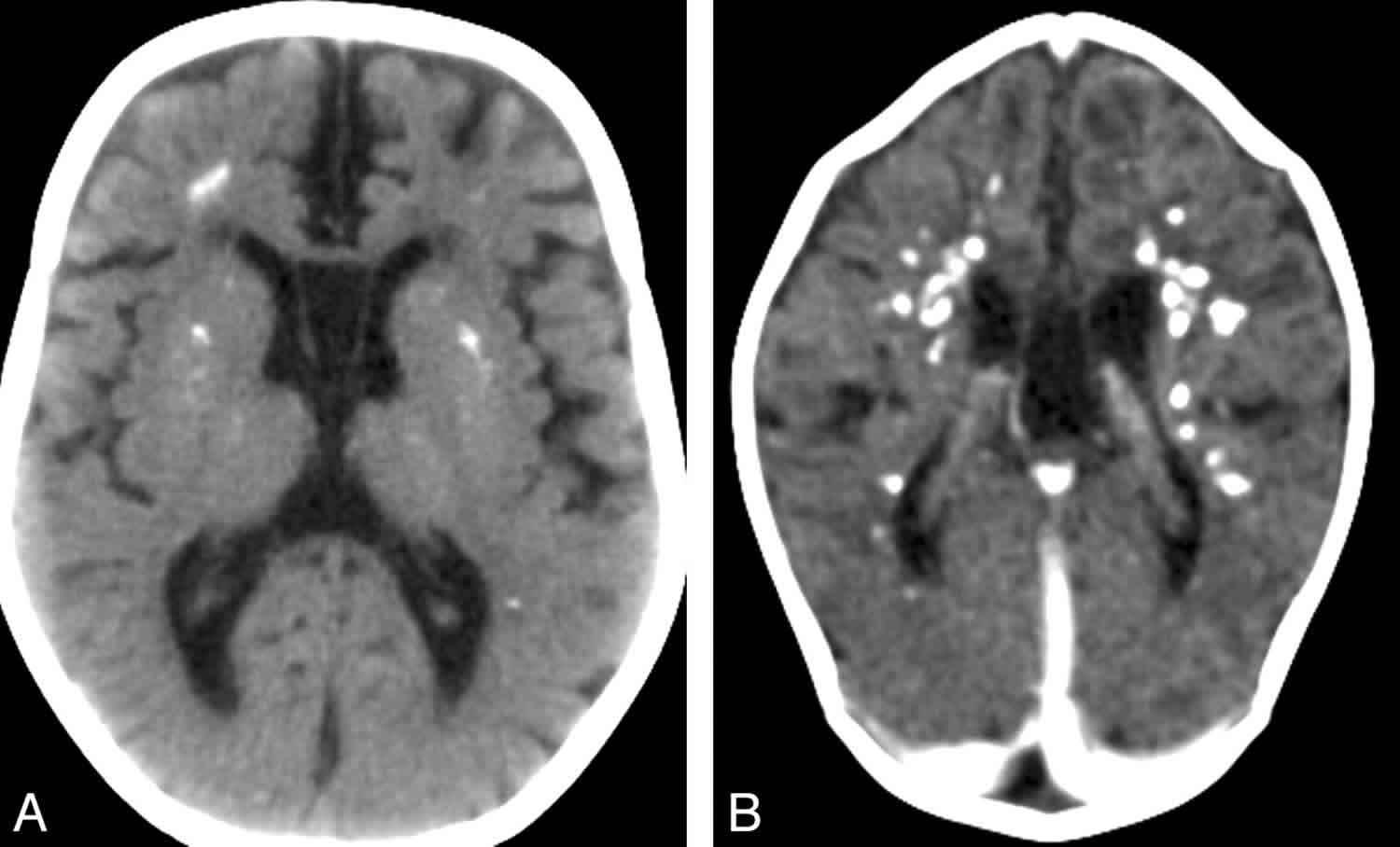
Footnote: Cerebral calcifications. (A) Axial nonenhanced CT image shows numerous punctuate calcifications within the basal ganglia and the cerebral white matter, a pattern typical in patients with Aicardi-Goutieres syndrome. (B) Contrast-enhanced CT scan shows large calcifications in the white matter. Although the CT examination was performed in the acute phase of the disease, no signs of contrast enhancement are seen. Atrophy, microcephaly, and areas of hypoattenuation in the periventricular white matter are also evident.
[Source 18) ]Figure 5. MRI characteristics in Aicardi-Goutieres syndrome

Footnote: MRI obtained in 2 patients with Aicardi-Goutieres syndrome at ages 4 months (upper row) and 11 months (lower row) in A and the second patient with images at 2 and 17 months in B. The images demonstrate early frontal and temporal lobe swelling (thin white arrow) that gives way to severe frontal and temporal lobe atrophy (thick white arrows), as well as the prominent global atrophy. Note also the T 2 dark signal abnormalities presumed to be calcifications present at 2 months and present at 17 months in the second patient (dashed arrows).
[Source 19) ]Supportive laboratory findings
Peripheral blood
- Positive interferon signature identified using quantitative PCR analysis of RNA/cDNA 20)
- Elevated liver enzymes
- Thrombocytopenia
Cerebrospinal fluid (CSF)
- Chronic CSF leukocytosis, defined as more than five lymphocytes/mm³ CSF.
- Typical values range from five to 100 lymphocytes/mm³ 21).
- A decrease in the number of lymphocytes occurs with time, although high cell counts may persist for several years.
- A normal cell count can be observed in the presence of elevated concentrations of IFN-α in the CSF even at an early stage of the disease 22).
- Increased interferon-alpha (IFN-α) activity in the CSF (normal: <2 IU/mL)
- Recorded IFN-α activity is usually highest in the early stages of the disease. The IFN-α CSF activity can normalize over the first three to four years of life 23).
- Recorded IFN-α activity is usually higher in CSF than in blood, where it may be normal.
- High IFN-α activity has been identified in fetal blood at 26 weeks’ gestation 24).
- Increased concentration of neopterin in the CSF 25)
- Levels are highest in the early stages of the disease and can normalize over time.
- Levels of the neurotransmitter metabolites 5HIAA, HVA, and 5MTHF are normal.
Is prenatal diagnosis possible?
Since Aicardi-Goutieres syndrome is, with rare exceptions, inherited as an autosomal recessive trait, most couples with an affected child have a 25% risk of the disease recurring in each future pregnancy. There exist rare cases in which it has been found to be inherited as an autosomal dominant trait (heterozygous TREX1, ADAR1 and IFIH1 mutations); in the majority of cases, these seem to be de novo mutations (except in rare families with mutations in ADAR1 and IFIH1 in which the mutation was transmitted by asymptomatic family members. These are real exceptions, to date found only for these two genes).
Today, genetic tests allow doctors to obtain a definite diagnosis of Aicardi-Goutieres syndrome in a large proportion of cases: however, it is important to note that diagnostic prenatal testing in at-risk pregnancies is possible only in families that already have an affected child, in whom the disease-causing gene has been identified. If the affected child’s mutation is known, then, in the event of a further pregnancy, the DNA of foetal cells obtained by chorionic villus sampling (at 10-12 weeks of gestation) or by amniocentesis (15-18 weeks of gestation) can be examined for the presence of the same mutation.
Establishing the diagnosis
The diagnosis of Aicardi-Goutieres syndrome is established in a proband with typical clinical findings and characteristic abnormalities on cranial CT (calcification of the basal ganglia and white matter) and MRI (leukodystrophic changes) and/or by the identification of biallelic pathogenic variants in ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, or TREX1; specific heterozygous autosomal dominant pathogenic variants in TREX1 and ADAR; or a variety of heterozygous autosomal dominant pathogenic variants in IFIH1.
Aicardi Goutieres syndrome treatment
Treatment of Aicardi-Goutieres syndrome is currently only symptomatic and supportive. Chest physiotherapy and treatment of respiratory complications; attention to diet and feeding methods to assure adequate caloric intake and avoid aspiration; management of seizures using drugs to control epilepsy.
The use of botulinum toxin and myorelaxant drugs to treat spasticity should be evaluated on a case by case basis in the context of the individual’s overall rehabilitation and care programme.
As regards the chilblain lesions, neither vasodilators nor immunosuppressants have been reported to show any real therapeutic efficacy in Aicardi-Goutieres syndrome and the treatment of these symptoms is limited to protecting the vulnerable parts from the cold and preventing infections that could complicate the situation.
Blau, in 2003, suggested that oral treatment with folinic acid might produce a general improvement of the clinical conditions in Aicardi-Goutieres syndrome patients presenting reduced folates in the CSF, but there are, as yet, no data in the literature confirming this hypothesis. In addition, a neurosurgical treatment to address the vascular complications (occlusive arterial and aneurysmal problems) has been proposed for patients with mutations in SAMHD1.
Surveillance: Patients must be regularly screened for the symptoms of the disease which are treatable, such as glaucoma or endocrine problems (e.g. diabetes or hypothyroidism). Monitoring for signs of diabetes insipidus in the neonatal period; repeat ophthalmologic examinations at least for the first few years of life to evaluate for evidence of glaucoma; monitoring for evidence of scoliosis, insulin-dependent diabetes mellitus, and hypothyroidism.
New therapies
Given the involvement and activation of the immune system in the pathogenesis of Aicardi-Goutieres syndrome, treatment with immunomodulatory therapies (prednisone + azathioprine, intravenous methylprednisolone + intravenous immunoglobulin (IVIG), methylprednisolone or IVIG alone) has been hypothesised and in some cases proposed. To date, however, these treatments have been used in an empirical manner and in a limited number of patients, and have not been clearly shown to modify the evolution of the disease, even in the rare cases treated in the early stages. It is, in fact, difficult to evaluate the efficacy of these treatments, considering the small number of patients involved and the differences between them, both in genotype and in disease stage at the time of treatment.
The progress in the understanding of the molecular mechanisms underlying the pathogenesis of Aicardi-Goutieres syndrome has led various authors to envisage new therapeutic strategies:
1) Biotech drugs
Given the suggested primary role of interferon in the pathogenesis of Aicardi-Goutieres syndrome, a treatment has been proposed based on blocking of interferon (IFN) activity through the use of monoclonal antibodies selective for different subtypes of IFN and for the type I IFN receptor (alpha and beta subunits). To date, this therapy has been used only on an experimental basis in SLE, and no data are available for Aicardi-Goutieres syndrome.
2) Immunomodulatory drugs
On the basis of the role of immune system dysregulation in the pathogenesis of Aicardi-Goutieres syndrome, it has been suggested that treatment could be based on the use of substances capable of destroying B cells (such as the monoclonal antibody rituximab) or of inhibiting autoreactive T cells (such as mycophenolate mofetil). However, these substances, often approved for paediatric use, can cause serious side effects and they have yet to be applied in therapeutic trials in the field of Aicardi-Goutieres syndrome.
3) Reverse transcriptase inhibitors
In view of the role of reverse transcriptase in the accumulation of DNA originating from endogenous RNA fragments (retroelements), normally metabolised by the proteins encoded by the genes that are mutated in Aicardi-Goutieres syndrome, it has been suggested that there might be a role in treatment of Aicardi-Goutieres syndrome for antiretroviral drugs (reverse transcriptase inhibitors, i.e. substances capable of interrupting the replication cycle both of retroviruses and of endogenous retroelements). The pharmacodynamics, safety and toxicity of these therapies are already well known, given that they are commonly prescribed to adults and children infected with HIV-1. At present, the only available data refer to an experimental trial in an animal model: Beck-Engeser showed that neurologically normal mice with the lethal TREX1-null phenotype could be kept alive using a combination of antiretroviral agents. Crow et al. have thus suggested that the next step could be to conduct a trial using drugs of this kind (even though their effectiveness could be limited by their difficulty crossing the blood-brain barrier) in order to test their applicability and safety in patients with Aicardi-Goutieres syndrome, and to assess whether this therapeutic approach is capable of “lowering” the “interferon signature” in those subjects in whom it can be considered, as we have seen, a biomarker of the disease, present even many years after the diagnosis.
It is indeed important to underline that the identification of an appropriate therapeutic strategy demands, in addition to a homogeneous sample of subjects, the use of easily quantifiable parameters of outcome (response to treatment), such as the interferon signature.
These new drugs, if applied in the early stage of the disease, could lead to an attenuation of the inflammatory process and thus a lessening of the tissue damage; instead, if applied in patients who are in an advanced stage of the disease and already show marked neurological impairment, they could result in some improvement of the clinical manifestations associated with the immune system dysregulation (e.g. chilblains). At present, there is no proof of the effectiveness of these possible therapeutic approaches, since the first pilot clinical trial is ongoing and has not yet provided results.
References [ + ]




{kind=link}