Contents
PTEN hamartoma tumor syndrome
PTEN hamartoma tumor syndrome refers to rare clinical syndromes characterized by germline mutations of the tumor suppressor PTEN that are characterized by multiple hamartomas 1). Hamartomas are benign (non cancer) growth made up of an abnormal mixture of cells and tissues normally found in the area of the body where the growth occurs. PTEN hamartoma tumor syndrome includes 2):
- Cowden syndrome – Cowden syndrome is a multiple hamartoma syndrome with a high risk for benign and malignant (cancerous) tumors of the thyroid, breast, and endometrium. Affected individuals usually have macrocephaly (large head size), trichilemmomas, and papillomatous papules, and present by the late 20s. The lifetime risk of developing breast cancer is 85%, with an average age of diagnosis between 38 and 46 years. The lifetime risk for thyroid cancer (usually follicular, rarely papillary, but never medullary thyroid cancer) is approximately 35%. The risk for endometrial cancer may approach 28%.
- Bannayan-Riley-Ruvalcaba syndrome – Bannayan-Riley-Ruvalcaba syndrome is a congenital disorder characterized by macrocephaly (large head size), intestinal hamartomatous polyposis (hamartomas of the intestines), lipomas, and pigmented macules of the glans penis.
- Proteus syndrome – Proteus syndrome is a complex, highly variable disorder involving congenital malformations and hamartomatous overgrowth of multiple tissues, as well as connective tissue nevi, epidermal nevi, and hyperostoses (overgrowth of the bones).
- Proteus-like syndrome – people with many of the signs and symptoms associated with Proteus syndrome, but who do not meet the diagnostic criteria.
Cowden syndrome was estimated to affect 1 in 200,000 individuals; this study was conducted just as PTEN hamartoma tumor syndrome was discovered. However, because the disorder is difficult to recognize, researchers believe it is under-diagnosed, making it difficult to determine its true frequency in the general population. Men and women are affected equally with PTEN hamartoma tumor syndrome. PTEN hamartoma tumor syndrome is not more commonly found in persons of a particular racial or ethnic group.
Fortunately most tumors that develop in persons with PTEN hamartoma tumor syndrome are benign,meaning they will not turn into a malignant cancer that can then metastasize, or spread, to other body parts. Persons with PTEN hamartoma tumor syndrome commonly develop benign growths, most of which are small, of the skin, tongue, and gums by adulthood. It has recently been discovered that colon polyps of various types, most of which have a low potential to develop into a malignancy, are seen in most adults who have an endoscopy (colonoscopy or upper). Benign breast lumps, thyroid nodules/goiter, and uterine fibroids are also common. Vascular malformations needing surgical intervention may also occur. A benign tumor of the cerebellum called Lhermitte-Duclos disease develops in a minority of adults with PTEN hamartoma tumor syndrome. Macrocephaly (larger than average head size) is common, and some children with PTEN hamartoma tumor syndrome are identified due to the presence of developmental delays and autism spectrum disorders. However, many persons with PTEN hamartoma tumor syndrome had no developmental challenges early in life and have successful careers as doctors, lawyers, or whatever other path they wished to follow.
For a person with PTEN hamartoma tumor syndrome, three different studies have found increased lifetime risks for specific cancer types 3). The study with the largest number of patients found the following cancer risks (to age 70): breast (85%), thyroid (35%), kidney (34%), uterus (28%), colorectal (9%) and melanoma (6%) 4). This study also recommends the following cancer screening recommendations:
- Breast (women): start high-risk breast screening (mammogram or MRI) plus clinical breast exam every 6 months starting at age 30
- Thyroid: annual ultrasound starting at age of diagnosis
- Kidney: imaging every two years starting at age 40
- Uterine: see a gynecologic oncologist starting at age 30 to discuss uterine cancer surveillance options
- Colon: baseline colonoscopy at age 35-40; follow-up dependent on number and type of polyps
- Skin: yearly dermatologic checks
Some patients wish to consider prophylactic surgeries, like mastectomy (breast) and hysterectomy (uterus) where at-risk tissue is removed prior to cancer development.
While some may develop hundreds of colon polyps, others may only develop a few. Similarly, while some persons have such severe thyroid disease that they require more frequent monitoring or even a thyroidectomy (thyroid removal) surgery, others do not. Given the differing needs of each person, screening and healthcare recommendations often vary from person to person. It can be helpful for persons with a complex condition like PTEN hamartoma tumor syndrome to have a “quarterback” who understands their management needs and helps coordinate visits and screenings with appropriate specialists; for some this quarterback may be their primary care provider, for others it is the genetic counselor or geneticist who made their diagnosis. Persons with PTEN hamartoma tumor syndrome are commonly connected with specialty physicians in the areas of high-risk breast care, endocrinology, gynecology-oncology, dermatology, and gastroenterology to help with their management.
PTEN hamartoma tumor syndrome is caused by changes (mutations) in the PTEN gene and is inherited in an autosomal dominant manner, this means that each child of an affected person has a 50% chance to inherit their PTENgene mutation and thus also have PTEN hamartoma tumor syndrome. It also means that their siblings, parents, and other relatives are at increased risk to share their specific PTEN mutation. Once a PTEN mutation is known to exist in a family, it is much cheaper for relatives to have testing for a known mutation as opposed to testing the entire PTEN gene. If you have PTEN hamartoma tumor syndrome, consider sharing your PTEN testing results with your relatives. If they have a copy of your test result, they can take it to any genetics provider, who will then be able to discuss testing with them and help them figure out if being tested is the right decision for them at this point in their lives.
PTEN hamartoma tumor syndrome treatment is based on the signs and symptoms present in each person 5).
Figure 1. PTEN hamartoma tumor syndrome
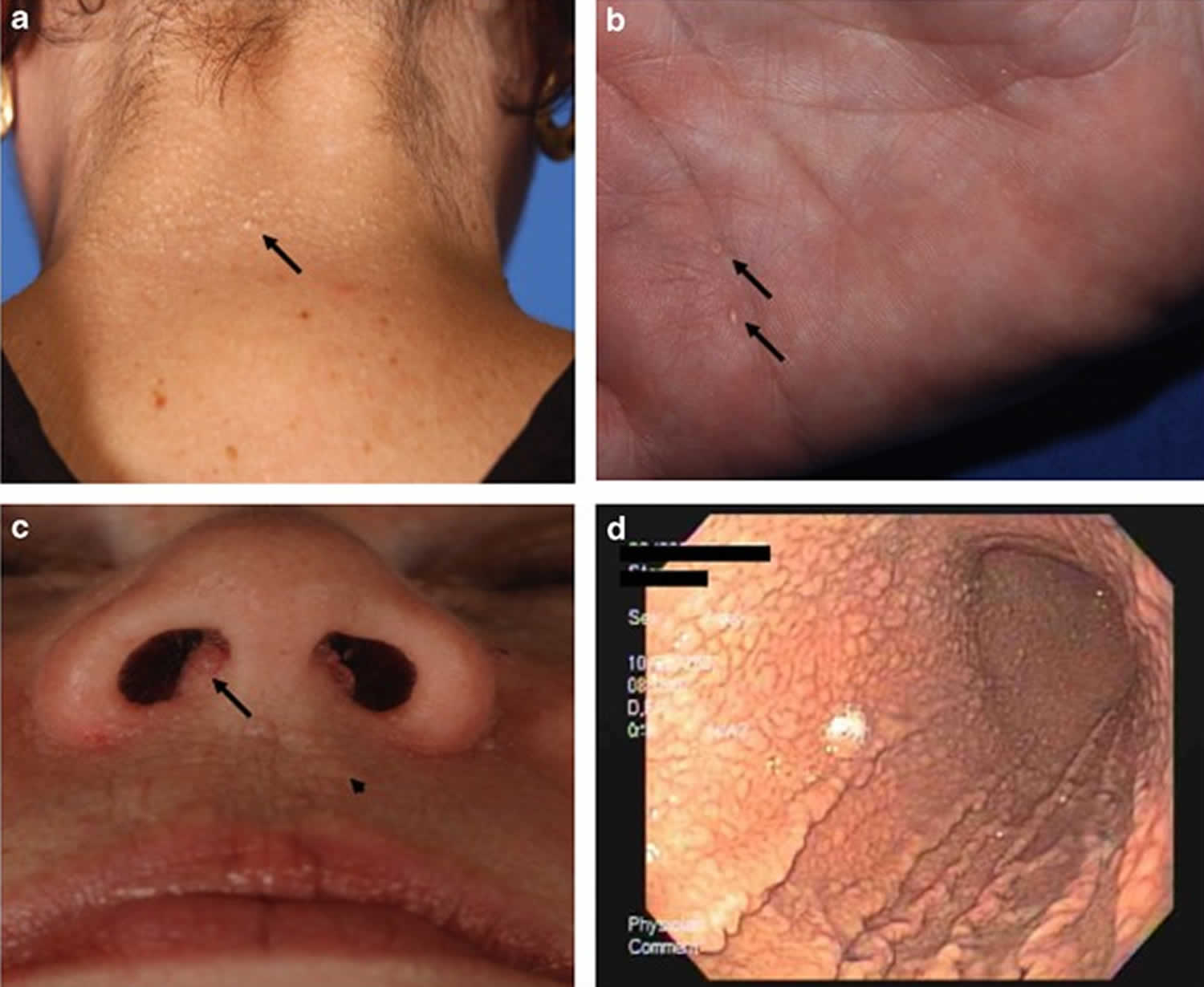
Footnote: Pathognomonic mucocutaneous features of Cowden syndrome. (a) Trichilemmomas on the nape of the neck of a subject with Cowden syndrome. (b) Palmar keratoses in a subject with Cowden syndrome. (c) Perioral papillomatous papules (arrow head) and nasal polyposis. (d) Gastric hamartomas as seen by endoscopy in a subject with Cowden syndrome.
PTEN hamartoma tumor syndrome causes
PTEN hamartoma tumor syndrome is caused by a germline mutation of PTEN, a tumor suppressor gene. PTEN stands for phosphatase tensin homologue. A tumor suppressor is a gene that slows down cell division, repairs damage to the DNA of cells, and tells cells when to die, a normal process called apoptosis. Mutations in a tumor suppressor gene often lead to cancer. The PTEN gene regulates the production of an enzyme (protein tyrosine phosphatase) which is believed to be important in stopping cell growth and starting apoptosis. Researchers believe that the PTEN gene plays a broad role in the development of human malignancies.
PTEN hamartoma tumor syndrome inheritance pattern
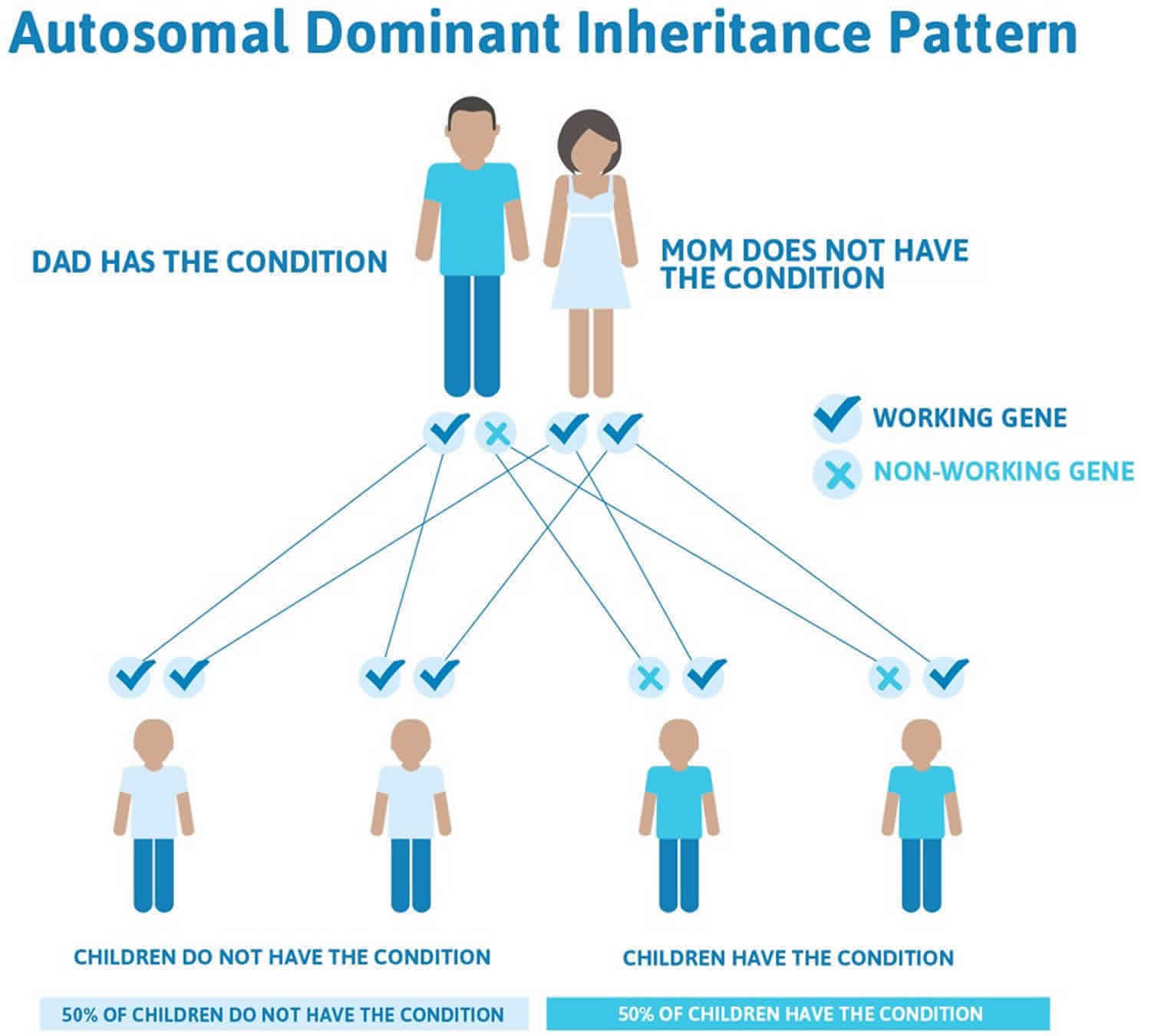
PTEN hamartoma tumor syndrome is inherited in an autosomal dominant pattern. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary to cause a particular disease. The abnormal gene can be inherited from either parent or can be the result of a mutated (changed) gene in the affected individual. The risk of passing the abnormal gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 2 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 2. PTEN hamartoma tumor syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
PTEN hamartoma tumor syndrome signs and symptoms
The primary findings in PTEN hamartoma tumor syndrome include increased risk for certain types of cancer, benign tumors and tumor-like malformations (hamartomas), and neurodevelopmental disorders. The symptoms of PTEN hamartoma tumor syndrome vary greatly from person to person and can develop at any age.
Cancer in PTEN hamartoma tumor syndrome
Previous data, which focused only on patients with a clinical diagnosis of Cowden syndrome without understanding whether an underlying PTEN mutation was present, estimated lifetime breast cancer risk to be 25-50% and risk for non-medullary thyroid cancer to be 10%. Risks for endometrial (uterine) and renal cell (kidney) cancer were thought to be increased, but an exact risk level was undetermined.
Current data focusing on patients known to have PTEN hamartoma tumor syndrome provide the following lifetime risk estimates, with the majority of diagnoses occurring after age 30:
Table 1. Cancer in PTEN hamartoma tumor syndrome
| Cancer | Lifetime Risk with PTEN hamartoma tumor syndrome (%) | Average Age at presentation |
| Breast | 85 | 40s |
| Thyroid | 35 | 30s-40s* |
| Renal Cell | 34 | 50s |
| Endometrial | 28 | 40s-50s |
| Colon | 9 | 40s |
| Melanoma | 6 | 40s |
Footnote: * Earliest age for thyroid cancer in PTEN hamartoma tumor syndrome is as early as 7 years old
Benign tumors in PTEN hamartoma tumor syndrome
Benign skin or oral lesions are very common and tend to appear in early adulthood. The most common types of benign skin lesions seen in PTEN hamartoma tumor syndrome include:
- Lipomas – benign fatty tumors which can appear just under the skin or elsewhere (breast area, GI tract)
- Acral keratosis – rough patches of skin most often seen on the extremities (arms, hands, legs, feet)
- Papillomatous skin papules – wart-like lesions which can appear anywhere, with feet and hands commonly being affected
- Mucosal papillomas – Benign overgrowth of tissue affecting the tongue, gums, or inside the nose
- Trichilemmomas – Benign tumor of the hair follicle
- Fibromas – another kind of overgrowth involving the skin and other connective tissue; may also affect tissue covering organs, such as the ovaries.
Gastrointestinal polyps are very common in adults with PTEN hamartoma tumor syndrome. Among patients who had undergone colonoscopy as part of their clinical care, >90% were found to have polyps with a mix of histological subtypes. The most common types of polyps found are hyperplastic or hamartomatous, which rarely develop into malignancy; however, adenomas, which may develop into a cancer, were also identified. Many polyps were very small and did not cause noticeable symptoms such as pain or rectal bleeding. Supported by this evidence, colorectal surveillance should be offered to any PTEN mutation carrier.
Benign breast, thyroid, and uterine lesions are also common in persons with PTEN hamartoma tumor syndrome. Some women have severe fibrocystic disease or changes which lead to multiple breast biopsies and complications with imaging. Multinodular goiter and Hashimoto’s thyroiditis may develop in children and adults. Uterine fibroids may appear and cause bleeding or discomfort to the extent that a hysterectomy is indicated without an underlying cancer diagnosis.
Vascular tumors, including hemangiomas, arteriovenous malformations, and developmental venous anomalies, have also been observed in patients with PTEN hamartoma tumor syndrome. Treatment of some lesions has been complicated by tendency for regrowth and scarring.
A small percentage of adults develop a rare tumor known as a cerebellar dysplastic gangliocytoma (Lhermitte-Duclos syndrome). Symptoms of Lhermitte-Duclos syndrome include increased intracranial pressure, impaired ability to coordinate voluntary movements (ataxia), and seizures. It is rare when a person with adult-onset Lhermitte-Duclos does not have an underlying PTEN mutation, and observing this tumor type is an automatic indicator for PTEN testing.
Neurodevelopmental concerns in PTEN hamartoma tumor syndrome
Macrocephaly (large head size) is found in 94% of measured patients with PTEN hamartoma tumor syndrome and can be a helpful screening tool to identify patients for PTEN testing. In most patients, large head size is caused by overgrowth of brain tissue. The head shape also tends to be longer than wide (dolicocephaly).
Autism and other developmental disorders, such as intellectual disability and developmental delays, have been observed in patients with PTEN hamartoma tumor syndrome. In previous case series, up to 17% of children presenting with macrocephaly and an autism spectrum disorder alone were found to have an underlying PTEN mutation.
PTEN hamartoma tumor syndrome diagnosis
A diagnosis of PTEN hamartoma tumor syndrome may be suspected based upon a thorough clinical evaluation, a detailed patient history and the presence of characteristic findings. Recently, a mutation risk calculator has been developed which can estimate the risk for adults to have a PTEN mutation based on their personal history characteristics; this tool is available online at (https://www.lerner.ccf.org/gmi/ccscore). The diagnosis can only be confirmed when a mutation of the PTEN gene is identified.
PTEN hamartoma tumor syndrome treatment
Individuals with PTEN mutations should undergo cancer surveillance and screening at the time of diagnosis as follows to enable healthcare providers to detect any tumors at the earliest, most treatable stages. Current suggested screening by age includes:
Pediatric (below age 18)
- Yearly thyroid ultrasound starting at the time of diagnosis
- Yearly skin check with physical examination
- Consider neurodevelopmental evaluation
Adults
- Monthly breast self-examination
- Yearly thyroid ultrasound and dermatologic evaluation
- Women: breast screening (at minimum mammogram) yearly beginning at age 30; MRI may also be incorporated
- Women: annual transvaginal ultrasound and/or endometrial biopsy beginning at age 30
- Colonoscopy beginning at age 35-40; frequency dependent on degree of polyposis identified
- Biannual (every other year) renal imaging (CT or MRI preferred) beginning at age 40
For patients with a family history of a particular cancer type, screening may be considered 5-10 years prior to the youngest diagnosis in the family. For example, a patient whose mother developed breast cancer at 30 may begin breast surveillance at age 25-30.
Additional treatment for PTEN hamartoma tumor syndrome is symptomatic and supportive. Various techniques may be used to treat the mucocutaneous symptoms of Cowden syndrome including topical agents (e.g., 5-fluorouracil), the use of extreme cold to destroy affected tissue (cryosurgery), the removal of tissue or growths by through a process called curettage, in which a surgical tool shaped like a spoon (curette) is used to scrape away affect tissue, or destroying affected tissue by exposing it to laser beams (laser ablation) 6). Cutaneous lesions should be excised only if malignancy is suspected or symptoms (e.g., pain, deformity, increased scarring) are significant. Genetic counseling may be of benefit for affected individuals and their families.
Prevention of primary manifestations
Some women at increased risk for breast cancer consider prophylactic mastectomy, especially if breast tissue is dense or if repeated breast biopsies have been necessary. Prophylactic mastectomy reduces the risk of breast cancer by 90% in women at high risk 7).
Note: The recommendation of prophylactic mastectomy is a generalization for women at increased risk for breast cancer from a variety of causes, not just from PTEN hamartoma tumor syndrome.
No direct evidence supports the routine use of agents such as tamoxifen or raloxifene in individuals with PTEN hamartoma tumor syndrome to reduce the risk of developing breast cancer. Physicians should discuss the limitations of the evidence and the risks and benefits of chemoprophylaxis with each individual. In addition, the clinician must discuss the increased risk of endometrial cancer associated with tamoxifen use in a population already at increased risk for endometrial cancer.
Bannayan-Riley-Ruvalcaba syndrome
Screening recommendations have not been established for Bannayan-Riley-Ruvalcaba syndrome. Given recent molecular epidemiologic studies, however, individuals with Bannayan-Riley-Ruvalcaba syndrome and a germline PTEN pathogenic variant should undergo the same surveillance as individuals with Cowden syndrome.
Individuals with Bannayan-Riley-Ruvalcaba syndrome should also be monitored for complications related to gastrointestinal hamartomatous polyposis, which can be more severe than in Cowden syndrome.
Proteus syndrome and Proteus-like syndrome
Although the observation of germline PTEN pathogenic variants in a minority of individuals who meet the clinical diagnostic criteria for Proteus syndrome and Proteus-like syndrome is relatively new, clinicians should consider instituting the Cowden syndrome surveillance recommendations for individuals with these disorders who have germline PTEN pathogenic variants.
Agents and circumstances to avoid
Because of the propensity for rapid tissue regrowth and the propensity to form keloid tissue, it is recommended that cutaneous lesions be excised only if malignancy is suspected or symptoms (e.g., pain, deformity) are significant.
References [ + ]




{kind=link}