
Contents
- Mitochondrial DNA depletion syndrome
- What is mitochondria
- Mitochondrial DNA depletion syndrome causes
- Mitochondrial DNA depletion syndrome signs and symptoms
- TK2-related myopathic mitochondrial DNA depletion syndrome
- SUCLA2 and SUCLG1-related encephalomyopathic mitochondrial DNA depletion syndrome
- RRM2B-related encephalomyopathic mitochondrial DNA depletion syndrome
- DGUOK-related hepatocerebral mitochondrial DNA depletion syndrome
- MPV17-related hepatocerebral mitochondrial DNA depletion syndrome
- POLG-related Hepatocerebral mitochondrial DNA depletion syndrome
- C10orf2-related hepatocerebral mitochondrial DNA depletion syndrome
- Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) disease
- Mitochondrial DNA depletion syndrome diagnosis
- Mitochondrial DNA depletion syndrome treatment
- Symptomatic management for mitochondrial DNA depletion syndrome
- Nutritional modulation in mitochondrial DNA depletion syndrome
- Cofactor use in mitochondrial DNA depletion syndrome
- Liver transplantation in mitochondrial DNA depletion syndrome
- Thymidine reduction in mitochondrial neurogastrointestinal encephalomyopathy disease
Mitochondrial DNA depletion syndrome
Mitochondrial DNA depletion syndrome are genetically and clinically a heterogeneous group of autosomal recessive mitochondrial disorders that are characterized by a severe reduction in mitochondrial DNA (mtDNA) content in affected tissues without mutations or rearrangements in the mitochondrial DNA (mtDNA) leading to impaired energy production in affected tissues and organs 1). An adequate amount of mtDNA (mitochondrial DNA) is required for the production of key subunits of mitochondrial respiratory chain complexes and therefore for energy production. Therefore, mitochondrial DNA (mtDNA) depletion results in organ dysfunction that is likely due to insufficient synthesis of respiratory chain components needed for adequate energy production 2).
Mitochondrial depletion syndrome are due to defects in mitochondrial DNA (mtDNA) maintenance caused by mutations in nuclear genes that function in either mitochondrial nucleotide synthesis (TK2, SUCLA2, SUCLG1, RRM2B, DGUOK, and TYMP) or mtDNA replication (POLG and C10orf2). Mitochondrial depletion syndrome are phenotypically heterogeneous and can affect a specific organ or a combination of organs and usually classified as myopathic (i.e. hypotonia, muscle weakness, bulbar weakness), encephalomyopathic (i.e. hypotonia, muscle weakness, psychomotor delay), hepatocerebral (i.e. hepatic dysfunction, psychomotor delay) or neurogastrointestinal (i.e gastrointestinal dysmotility, peripheral neuropathy). Additional phenotypes include fatal infantile lactic acidosis with methylmalonic aciduria, spastic ataxia (early-onset spastic ataxia-neuropathy syndrome), and Alpers syndrome.
Myopathic mitochondrial depletion syndrome, caused by mutations in TK2 gene encoding thymidine kinase 2 are among the most common causes of mitochondrial depletion syndrome, usually present before the age of 2 years with hypotonia and muscle weakness, with about 200 patients that have been reported by two groups, Hirano’s 3) and Wang’s 4). Most of the patients with TK2 mutations have an infantile onset form, presenting at or soon after birth with generalized weakness, respiratory insufficiency, and death at 1 to 3 years of age 5). Severe skeletal muscle mitochondrial DNA (mtDNA) depletion is commonly observed. Lately, Michio Hirano and Caterina Garone have developed a life-saving therapeutic approach for these infants. The therapy provided oral deoxynucleosides (the substrates of the TK2 enzyme) and showed efficacy in ameliorating mtDNA depletion and increased lifespan in mice 6). Up to today, this treatment has been used in 16 patients worldwide under compassionate use, the exiting results obtained in this cohort will lead to a forthcoming clinical trial.
Encephalomyopathic mitochondrial depletion syndrome, caused by mutations in SUCLA2, SUCLG1, or RRM2B, typically present during infancy with hypotonia and pronounced neurological features.
Hepatocerebral mitochondrial depletion syndrome, caused by mutations in DGUOK, MPV17, POLG, or C10orf2, commonly have an early-onset liver dysfunction and neurological involvement.
Finally, TYMP mutations have been associated with mitochondrial neurogastrointestinal encephalopathy disease that typically presents before the age of 20 years with progressive gastrointestinal dysmotility and peripheral neuropathy.
Overall, mitochondrial depletion syndrome are severe disorders with poor prognosis in the majority of affected individuals. No efficacious therapy is available for any of these disorders. Affected individuals should have a comprehensive evaluation to assess the degree of involvement of different systems. Treatment is directed mainly toward providing symptomatic management. Nutritional modulation and cofactor supplementation may be beneficial. Liver transplantation remains controversial. Finally, stem cell transplantation in neurogastrointestinal encephalopathy disease shows promising results 7).
What is mitochondria
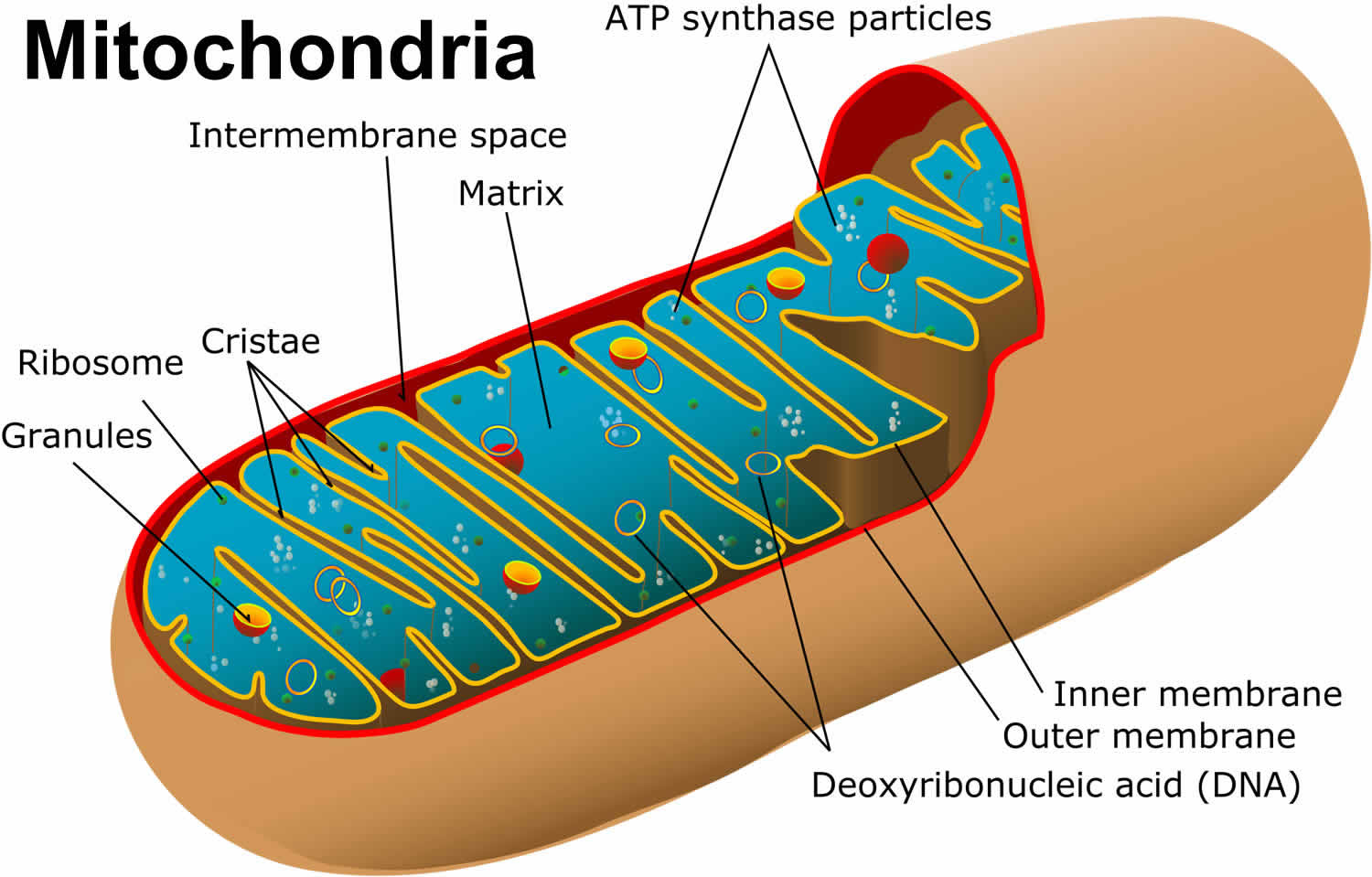
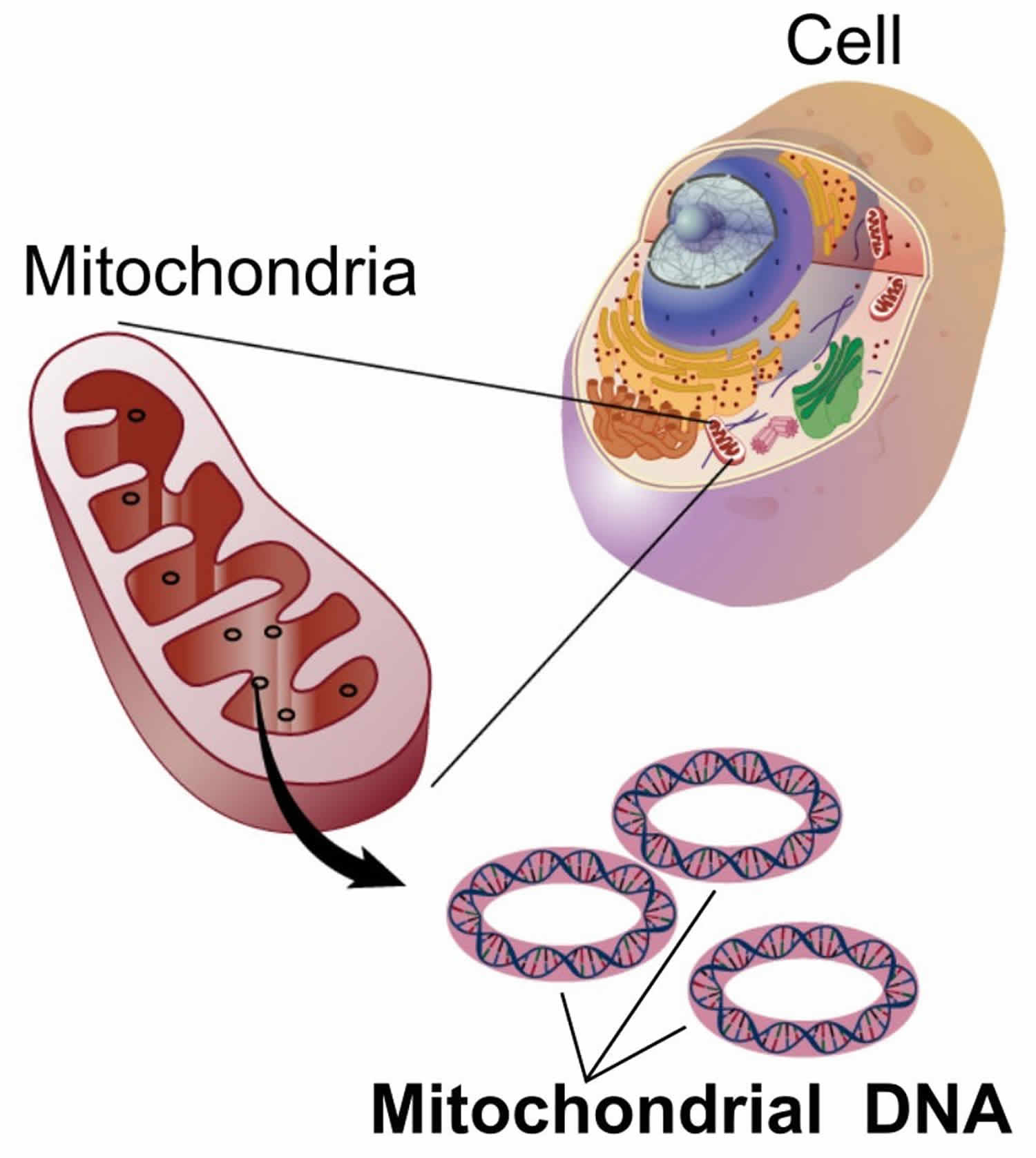
You have mitochondria present in every cell of your body except red blood cells. Mitochondria are membrane-bound cell organelles (mitochondrion, singular) that generate most of the chemical energy needed to power the cell’s biochemical reactions. The mitochondria in the cells throughout your body are responsible for creating more than 90% of the energy needed by your body to sustain life and support organ function. When mitochondria fail, less and less energy is generated within the cell. Cell injury and even cell death follow. If this process is repeated throughout the body, whole organ systems begin to fail – people get sick, and even die. Chemical energy produced by the mitochondria is stored in a small molecule called adenosine triphosphate (ATP). Mitochondria contain their own small chromosomes. Generally, mitochondria, and therefore mitochondrial DNA, are inherited only from the mother. Problems with mitochondria, the structures that produce energy for all cells, have been linked to the development of Parkinson’s disease.
Mitochondria play a fundamental role in cell physiology; mitochondria organelles are involved in a variety of processes, including bioenergetics, various metabolic pathways, including crucial anabolic and catabolic reactions, such as ATP (adenosine triphosphate) synthesis, the tricarboxylic acid cycle (citric acid cycle or Kreb cycle), and biosynthetic processes, and govern fundamental cellular actions, including proliferation, immunity, and autophagy. Mitochondrial damage and malfunction have been related to the pathogenesis of a large number of human pathologies, such as mitochondrial diseases, neurodegenerative diseases, cancer, cardiovascular diseases, metabolic disorders, and aging. The participation of mitochondria in the redox equilibrium and redox signaling of the cell is also pivotal. Modification of the redox state and increased reactive oxygen species (ROS) production within mitochondria have major consequences for both mitochondrial and extramitochondrial processes and, ultimately, modulate fundamental cellular phenomena such as autophagy and apoptosis.
In people with mitochondrial disease, the parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected 8). Based upon recent epidemiological studies, mitochondrial disorders affect at least 1 in 8000 of the general population 9). Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Figure 1. Mitochondria cell
Mitochondrial DNA depletion syndrome causes
Mitochondrial DNA depletion syndrome are due to defects in mtDNA maintenance caused by mutations in nuclear genes, which function in either maintaining the mitochondrial deoxyribonucleoside triphosphate (dNTP) pool – TK2 (thymidine kinase 2), DGUOK (deoxyguanosine kinase), SUCLA2 [adenosine diphosphate (ADP)-forming succinyl CoA ligase beta subunit], SUCLG1 [guanosine diphosphate (GDP)-forming succinyl CoA ligase alpha subunit], RRM2B (ribonucleotide reductase M2 B subunit), and TYMP (thymidine phosphorylase) or by mutations in genes associated with mtDNA replication [POLG (DNA polymerase gamma) and C10orf2 (Twinkle)]; therefore, mutations in these genes result in insufficient mtDNA synthesis to keep up with mtDNA turnover and segregation to daughter cells during cell divisions resulting in reduction of mtDNA content 10). The function of the MPV17 gene remains unclear (Figure 2).
Figure 2. Mitochondrial DNA depletion syndrome
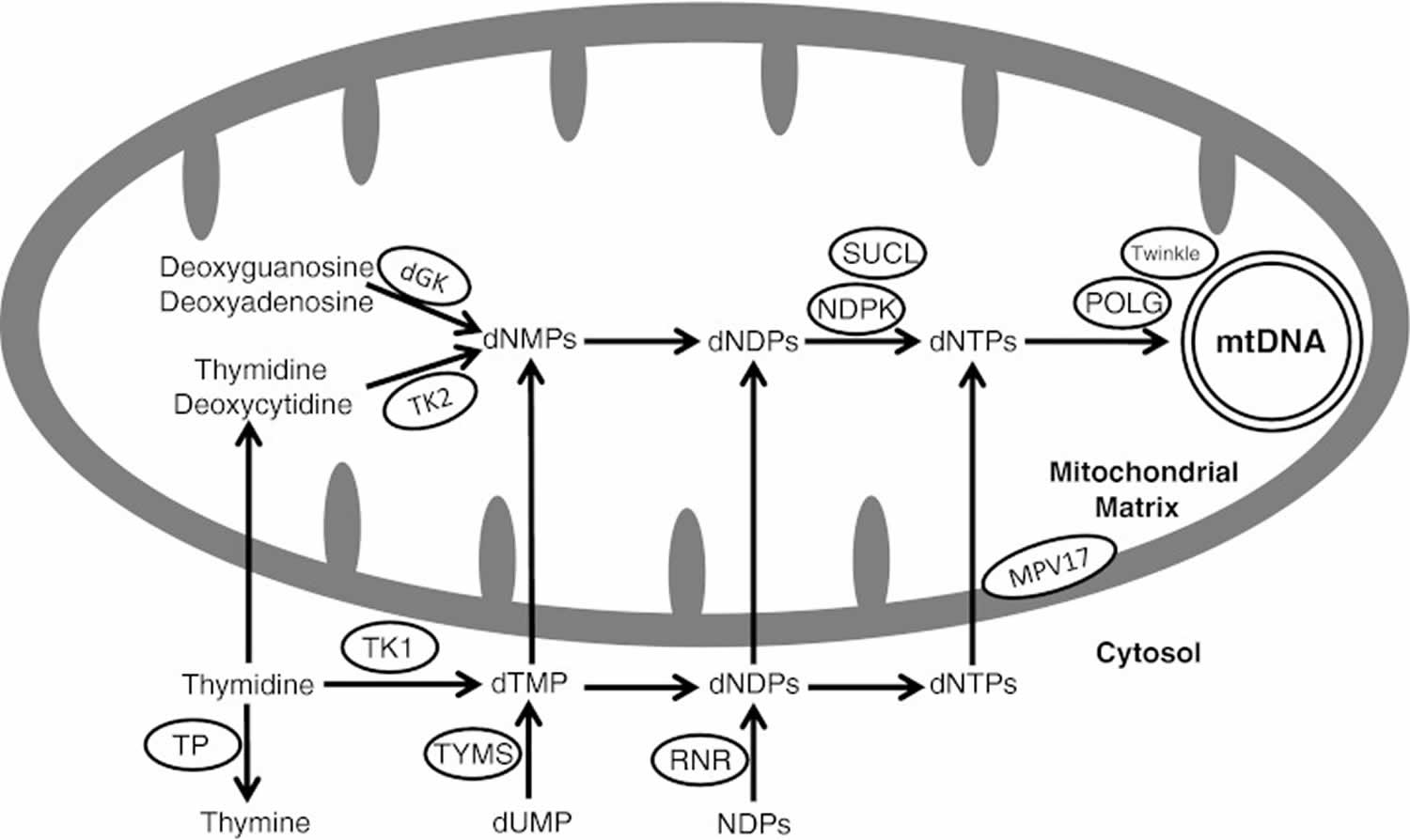
Footnote: Schematic presentation of protein involved in mitochondrial nucleotide pools maintenance and mitochondrial DNA replication. TK2 = mitochondrial thymidine kinase 2 (encoded by TK2 gene); dGK = mitochondrial deoxyguanosine kinase (encoded by the DGUOK gene); SUCL = succinyl CoA ligase (SUCL is composed of an alpha subunit, encoded by SUCLG1 and a beta subunit, encoded by either SUCLA2 or SUCLG2); NDPK = nucleoside diphosphate kinase; POLG = DNA polymerase gamma (POLG is a heterotrimer enzyme composed of one catalytic subunit encoded by POLG and two accessory subunits encoded by POLG2); TP = thymidine phosphorylase (encoded by TYMP gene); RNR = ribonucleotide reductase (RRM2B encodes the p53-inducible small subunit (p53R2) of the RNR); dNMP = deoxynucleoside monophosphate; dNDP = deoxynucleoside diphosphate; dNTP = deoxynucleoside triphosphate; NDP = nucleoside diphosphate; dTMP = deoxythymidine monophosphate; TK1 = cytosolic thymidine kinase 1; TYMS = thymidylate synthase. The twinkle protein is encoded by C10orf2 and the MPV17 by the MPV17 gene
[Source 11) ]Defects in maintaining mitochondrial nucleotide pool
Unlike nuclear DNA, which replicates with each cell division, mitochondrial DNA (mtDNA) replicates continuously and independently of cell division. Deoxyribonucleoside triphosphates (dNTPs) can be synthesized via either the de novo pathway, which is cell cycle-regulated, thereby operative only in S-phase cells or the salvage pathway in which deoxyribonucleoside triphosphates are produced by utilizing pre-existing deoxynucleosides to synthesize DNA precursors. As mtDNA synthesis is continuous throughout the cell cycle, the salvage pathway becomes essential for mtDNA maintenance. TK2, DGUOK, SUCLA2, SUCLG1, RRM2B, and TYMP encode proteins that maintain the mitochondrial deoxyribonucleoside triphosphate pool mainly through salvage pathways; therefore, mutations in any of these genes result in depleting the mitochondria from DNA building blocks with subsequent mtDNA depletion.
Mitochondrial thymidine kinase 2 (TK2) is encoded by the nuclear gene TK2 and plays an essential role in the pyrimidine nucleoside salvage pathway 12). It mediates the first, and rate-limiting, step in the phosphorylation of pyrimidine nucleosides in the mitochondrial matrix. Mitochondrial deoxyguanosine kinase is encoded by the nuclear gene DGUOK and is essential for the purine nucleoside salvage pathway as it mediates the first step in the phosphorylation of purine nucleosides in the mitochondrial matrix 13). Mutations in TK2 or DGUOK result in impaired synthesis of mitochondrial deoxyribonucleoside triphosphates (dNTPs), the building blocks for mtDNA, leading to decreased mtDNA amount and mtDNA depletion.
SUCLA2 and SUCLG1 encode subunits of succinyl CoA ligase (SUCL). SUCL is a mitochondrial tricarboxylic acid cycle enzyme that catalyzes the reversible conversion of succinyl-CoA and ADP or GDP to succinate and adenosine triphosphate or guanosine triphosphate. SUCL is composed of an alpha subunit, encoded by SUCLG1 and a beta subunit, encoded by either SUCLA2 or SUCLG2. The alpha subunit forms a heterodimer with either of its beta subunits, resulting in an ADP-forming SUCL and a GDP-forming SUCL, respectively. SUCL also forms a complex with the mitochondrial nucleoside diphosphate kinase, and the lack of this complex formation in SUCL deficiency has been suggested to disturb the kinase function, resulting in decreased mtDNA synthesis leading to mtDNA depletion 14).
RRM2B encodes the p53-inducible small subunit (p53R2) of ribonucleotide reductase, a cytosolic enzyme that catalyzes the terminal step of de novo synthesis of deoxyribonucleoside by direct reduction of ribonucleoside diphosphates to their corresponding deoxyribonucleoside diphosphates. The p53R2 is expressed in post-mitotic cells and therefore has a key function in the maintenance of deoxyribonucleoside triphosphate pools for mtDNA synthesis 15).
TYMP encodes thymidine phosphorylase (TP), which is a cytosolic enzyme that catalyzes the conversion of thymidine to thymine and deoxyuridine to uracil, and is therefore essential for the nucleotide salvage pathway. Low thymidine phosphorylase activity results in the accumulation of thymidine and deoxyuridine, leading to an imbalance of cytosolic deoxyribonucleoside triphosphate pools. Because the mitochondrial deoxyribonucleoside triphosphate pool relies, in part, on deoxyribonucleoside triphosphate imported from the cytosol, an imbalanced cytosolic deoxyribonucleoside triphosphate pool can lead to an imbalanced mitochondrial deoxyribonucleoside triphosphate pool that can impair mtDNA synthesis 16).
Defects in mtDNA replication
POLG encodes the catalytic subunit of DNA polymerase gamma, which is a heterotrimer enzyme composed of one catalytic subunit encoded by POLG and two accessory subunits encoded by POLG2 that assist in binding and processing the synthesized DNA. DNA polymerase gamma is required for mtDNA synthesis as it is the only DNA polymerase in humans that allows for replication and repair of mtDNA 17). The twinkle protein, encoded by C10orf2, serves the important function of a DNA helicase that is required for DNA replication 18). Therefore, POLG and C10orf2 are essential for mtDNA replication and mutations in these genes result in insufficient mtDNA synthesis to keep up with mtDNA turnover and segregation to daughter cells during cell divisions, resulting in a reduction of mtDNA content and mtDNA depletion.
mtDNA depletion caused by defects in a protein of unknown function
MPV17 encodes the MPV17 protein, an inner mitochondrial membrane protein whose function and role in the pathogenesis of mtDNA depletion are as yet unknown. It has been suggested that MPV17 plays a role in controlling mtDNA maintenance and oxidative phosphorylation activity in mammals and yeast 19). A dysfunctional MPV17 protein caused by MPV17 mutations impairs mtDNA maintenance and can cause mtDNA depletion.
Mitochondrial DNA depletion syndrome signs and symptoms
Mitochondrial DNA depletion syndrome are phenotypically heterogeneous and may affect either a specific organ or a combination of organs, including muscle, liver, brain, and kidney. Clinically, mitochondrial DNA depletion syndrome are usually classified as 1 of 4 forms 20):
- a myopathic form associated with mutations in TK2;
- an encephalomyopathic form associated with mutations in SUCLA2, SUCLG1, or RRM2B;
- a hepatocerebral form associated with mutations in DGUOK, MPV17, POLG, or C10orf2; and
- a neurogastrointestinal form associated with mutations in TYMP.
Table 1. Clinical phenotypes of different mitochondrial DNA depletion syndromes
| Mitochondrial DNA depletion syndromes | Age of onset | Common clinical features |
|---|---|---|
| Myopathic | ||
| TK2-related | Infancy—early childhood | Hypotonia and muscle weakness, facial weakness, bulbar weakness (dysarthria and dysphagia), elevated serum creatine phosphokinase |
| Encephalomyopathic | ||
| SUCLA2– and SUCLG1-related | Infancy | Hypotonia and muscle weakness, psychomotor delay, scoliosis/kyphosis, abnormal movement disorders (dystonia, athetoid, or choreiform), sensorineural hearing impairment, epilepsy, growth retardation, lactic acidosis, elevated methylmalonic acid in urine and plasma, cortical atrophy and basal ganglia involvement in neuroimaging |
| RRM2B-related | Neonatal—infancy | Hypotonia and muscle weakness, psychomotor delay, microcephaly, sensorineural hearing loss, failure to thrive, lactic acidosis |
| Hepatocerebral | ||
| DGUOK-related | Neonatal | Hepatic dysfunction, psychomotor delay, hypotonia, rotary nystagmus developing into opsoclonus, lactic acidosis, hypoglycemia |
| MPV17-related | Infantile—childhood | Hepatic dysfunction, psychomotor delay, hypotonia, peripheral neuropathy, lactic acidosis, hypoglycemia, leukoencephalopathy in neuroimaging |
| POLG-related | Early childhood | Hepatic dysfunction, epilepsy, psychomotor delay, ataxia, neuropathy, hyporeflexia and hypotonia evolving into spastic paraparesis, stroke or stroke-like episodes, myoclonus, choreoathetosis, parkinsonism, nystagmus, somnolence, irritability, cortical visual loss, and sensorineural hearing impairment, generalized brain atrophy in neuroimaging |
| C10orf2-related | Neonatal—infancy | Hepatic dysfunction, psychomotor delay, epilepsy, peripheral neuropathy, hypotonia, ophthalmoplegia, nystagmus, athetosis, ataxia, sensorineural hearing impairment, lactic acidosis, cerebellar cortical atrophy in neuroimaging |
| Neurogastrointestinal | ||
| TYMP-related | Late childhood—adolescence | Gastrointestinal dysmotility, weight loss, peripheral neuropathy, ptosis, ophthalmoplegia, elevated thymidine and deoxyuridine in plasma, leukoencephalopathy in neuroimaging |
To date, approximately 50 affected individuals have been reported with TK2-related mitochondrial DNA depletion syndrome. The clinical presentation of TK2-related mitochondrial DNA depletion syndrome is variable, with a broad phenotype. Initial development is typically normal and the majority of affected children present before the age of 2 years with gradual onset of hypotonia, generalized fatigue, decreased physical stamina, proximal muscle weakness, and feeding difficulty. Some patients develop facial weakness and bulbar weakness, including dysarthria and dysphagia. Hypotonia and weakness is observed in all patients and previously acquired motor skills are lost. However, cognitive function is typically spared 21).
Although TK2-related mitochondrial DNA depletion syndrome has been thought to be associated with a purely myopathic form, other organ system involvements have been reported, including an encephalomyopathic presentation with hypotonia, weakness, epilepsy, and microcephaly 22), and hepatic involvement with hepatomegaly and elevated transaminases accompanied by mtDNA depletion in muscle and liver [19]. Other, less common, presentations include spinal muscular atrophy-like presentation 23) and chronic progressive external ophthalmoplegia with proximal muscle weakness 24). Milder presentations have been reported and include late onset proximal muscle weakness 25), adult-onset progressive myopathy 26) and sensorineural hearing loss 27).
Serum creatine phosphokinase concentration is usually elevated and electromyography (EMG) usually shows non-specific myopathic changes. Histopathological findings on skeletal muscle include prominent variance in fiber size, sarcoplasmic vacuoles, and increased connective tissue. Ragged red fibers are present. Succinate dehydrogenase activity is increased, whereas cytochrome c oxidase activity is low, or absent. Electron microscopy shows abnormal mitochondria with circular cristae. mtDNA content is typically severely reduced in muscle tissue. Electron transport chain (ETC) activity assays in skeletal muscle typically show decreased activity of multiple complexes with complex I, I + III, and IV being the most affected 28).
Typically, muscle weakness rapidly progresses leading to respiratory failure and death within a few years of onset. The most common cause of death is pulmonary infection. Only a few patients have survived to late childhood and adolescence.
Nearly 20 individuals have been reported with SUCLA2-related mitochondrial DNA depletion syndrome. Mutations in SUCLG1 have been reported less frequently with a similar phenotype to that observed in SUCLA2-related mitochondrial DNA depletion syndrome 29). Affected infants present with hypotonia typically before the age of 6 months. All affected children develop hypotonia, muscle atrophy, and psychomotor delay. Other frequent manifestations include progressive scoliosis or kyphosis, abnormal movements, including dystonia and athetoid or choreiform movements, feeding difficulty, gastroesophageal reflux, sensorineural hearing impairment, postnatal growth retardation, and respiratory insufficiency that can result in frequent pulmonary infections. Other, less common, manifestations include hyperhidrosis, strabismus, ptosis, and epilepsy presenting with either infantile spasms or generalized convulsions. Urine organic acids analysis consistently shows elevated methylmalonic acid. Similarly, plasma methylmalonic acid concentration is elevated. Lactate is elevated in both plasma and cerebrospinal fluid (CSF) in most affected individuals. EMG may reveal findings suggestive of motor neuron involvement, whereas neuroimaging may show cortical atrophy, bilateral basal ganglia involvement, and delayed myelination. Histopathological findings on skeletal muscle include increased fiber variability, increased number of mitochondria, and extensive intracellular fat accumulation. ETC activity assays in muscle typically show a combined deficiency of respiratory complex I, III, and IV, with normal complex II activity. Quantitation of mtDNA shows a decreased mtDNA content in muscle. Prognosis is poor, with most affected children dying in childhood, most commonly from an intercurrent infection 30).
To date, RRM2B mutations have been reported in about 15 infants with severe encephalomyopathic mitochondrial DNA depletion syndrome that is associated with early-onset (neonatal or infantile), multi-organ presentation, and mortality during infancy. Affected individuals typically present during the first months of life with hypotonia, lactic acidosis, failure to thrive, tubulopathy, microcephaly, psychomotor delay, sensorineural hearing loss, and profound mtDNA depletion in muscle. The disease progresses rapidly, leading to death in few months 31).
RRM2B mutations have also been reported to cause a mitochondrial neurogastrointestinal encephalopathy (MNGIE)-like phenotype with mtDNA depletion 32) and autosomal-dominant progressive external ophthalmoplegia (PEO) with multiple mtDNA deletions 33).
Approximately 100 individuals have been reported with DGUOK-related mitochondrial DNA depletion syndrome, which can present in two forms: multi-organ disease in neonates and isolated hepatic disease later in infancy or childhood 34). The majority of affected individuals have a neonatal-onset multi-organ illness that presents with lactic acidosis and hypoglycemia in the first week of life. Within weeks of birth, all infants develop hepatic disease and neurologic dysfunction. Severe myopathy, developmental regression, and typical rotary nystagmus developing into opsoclonus are also seen. Cholestasis is prominent early in the clinical course. Liver involvement may cause neonatal- or infantile-onset liver failure that is generally progressive with ascites, edema, and coagulopathy. A minority of affected individuals present initially in infancy or childhood with isolated hepatic disease, occasionally following a viral illness. Affected individuals with this form may develop mild hypotonia and renal involvement manifesting as proteinuria and aminoaciduria 35). More recently, DGUOK mutations have been reported in a neonate with clinical and autopsy findings consistent with neonatal hemochromatosis and mtDNA depletion 36), and in individuals with adult-onset mitochondrial myopathy and mtDNA multiple deletions in skeletal muscle 37).
The majority of affected newborns with the multi-organ form of the disease show elevated serum concentration of tyrosine or phenylalanine on newborn screening 38). Findings of intrahepatic cholestasis typically include elevations in serum concentrations of liver transaminases, gamma-glutamyltransferase (GGT), and conjugated hyperbilirubinemia. Increased serum concentration of ferritin is observed in a large number of affected infants. mtDNA content is reduced in liver and muscle 39). Electron transport chain (ETC) activity in liver typically shows a combined deficiency of complexes I, III, and IV 40). Liver histopathology typically reveals microvesicular cholestasis, but may show bridging fibrosis, giant cell hepatitis, or cirrhosis. Liver electron microscopy may reveal an increase in the number of mitochondria and is commonly associated with abnormal cristae 41).
Hepatic dysfunction is progressive in the majority of individuals with both forms of DGUOK-related mitochondrial DNA depletion syndrome and is the most common cause of death. Hepatocellular carcinoma has also been reported in 1 patient. For children with the multi-organ form, liver transplantation provides no survival benefit 42).
MPV17-related hepatocerebral mitochondrial DNA depletion syndrome, an infantile-onset disorder, can present with a spectrum of combined hepatic, neurologic, and metabolic manifestations. Approximately 30 affected individuals have been reported with MPV17-related hepatocerebral mitochondrial DNA depletion syndrome 43). Of note, among those confirmed cases are individuals with Navajo neurohepatopathy who were found to have homozygous p.Arg50Gln mutations in MPV17. Navajo neurohepatopathy, a disorder prevalent in the Native American Navajo population, has the manifestations of MPV17-related hepatocerebral mitochondrial DNA depletion syndrome, as well as painless fractures, acral mutilation, and corneal anesthesia, ulceration, and scarring 44).
Affected individuals typically present with manifestations of liver dysfunction, including jaundice, cholestasis, and coagulopathy. Infancy is the typical age of onset; however, individuals homozygous for the p.Arg50Gln mutation may present later in childhood 45). In the vast majority of affected individuals, liver disease progresses to liver failure typically during infancy or early childhood. Hepatomegaly and liver cirrhosis occur in some affected individuals. Hepatocellular carcinoma has also been reported in 2 affected individuals. The vast majority of affected individuals exhibited neurologic manifestations, including developmental delay, hypotonia, muscle weakness, and motor and sensory peripheral neuropathy. Some affected individuals presented with psychomotor delays during early infancy, while others had normal development early in life followed by loss of motor and cognitive abilities later in infancy or early childhood. Less frequent neurologic manifestations include epilepsy, ataxia, dystonia, microcephaly, cerebrovascular infarction, and subdural hematoma. Failure to thrive is one of the common manifestations, although some children have normal growth, especially early in the course of the disease. The vast majority of affected individuals have metabolic derangements, including lactic acidosis and hypoglycemia, which typically presents during the first 6 months of life. Less frequent manifestations include renal tubulopathy, hypoparathyroidism, and gastrointestinal dysmotility that manifests as gastroesophageal reflux, cyclic vomiting, and diarrhea. Corneal anesthesia and ulcers were reported in individuals homozygous for the mutation p.Arg50Gln 46).
More recently, MPV17 mutations have been reported in adult presentation of neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle indicating that MPV17 mutations are associated with an evolving broader phenotype 47).
Affected infants demonstrate elevated transaminases and GGT, and hyperbilirubinemia. Liver histopathology may show cholestasis and cirrhosis. Neuroimaging may show white matter abnormalities (leukoencephalopathy). mtDNA content is severely and consistently reduced in liver tissue, and can also be reduced in muscle tissue. Electron transport chain (ETC) activity assays in liver and muscle tissue typically show decreased activity of multiple complexes with complex I or I + III being the most affected 48).
Liver disease typically progresses to liver failure in affected children and liver transplantation remains the only treatment option for liver failure. Approximately half of affected children reported did not undergo liver transplantation and died because of progressive liver failure—the majority during infancy or early childhood. A few children were reported to survive without liver transplantation 49).
POLG-related disorders present a continuum of broad and overlapping phenotypes presenting from early childhood to late adulthood. The clinical phenotypes of POLG-related disorders include autosomal recessive and dominant adult-onset progressive external ophthalmoplegia 50), myoclonic epilepsy, myopathy, sensory ataxia (MEMSA) syndrome 51), ataxia-neuropathy spectrum including mitochondrial recessive ataxia syndrome (MIRAS), and sensory ataxia, neuropathy, dysarthria, ophthalmoplegia (SANDO) syndrome 52), and hepatocerebral mitochondrial DNA depletion syndrome (Alpers-Huttenlocher syndrome) 53). More recently, POLG mutations were identified in individuals with clinical features of mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), but no leukoencephalopathy 54).
The incidence of Alpers-Huttenlocher syndrome has been estimated to be ~1:50,000 55). It is the most severe phenotype associated with POLG mutations and characterized by a progressive encephalopathy with intractable epilepsy and psychomotor delay, neuropathy, and hepatic failure. Affected individuals usually present between the age of 2 and 4 years with seizures (focal, generalized, myoclonic, epilepsia partialis continua, or status epilepticus), headaches that are typically associated with visual sensations or visual auras, hypotonia, and psychomotor regression. Early in the disease course areflexia and hypotonia are present and later followed by spastic paraparesis that evolves over months to years, leading to psychomotor regression. All affected individuals develop neuropathy, ataxia, and loss of cognitive function, including concentration, language skills, and memory. Affected individuals may also develop stroke and stroke-like episodes, myoclonus, choreoathetosis, parkinsonism, nystagmus, somnolence, irritability, loss of normal emotional responses, depression, cortical visual loss, and sensorineural hearing loss. Neurologic signs and symptoms may worsen during infections or other stressful situations. Affected individuals develop liver dysfunction with elevated transaminases, hypoalbuminemia, coagulopathy, hypoglycemia, and hyperammonemia. Liver involvement can progress rapidly to end-stage liver failure within a few months. CSF protein is generally elevated. Neuroimaging may show gliosis and generalized brain atrophy. Liver histology may demonstrate macro- and microvesicular steatosis, centrilobular necrosis, fibrosis, cirrhosis, bile duct proliferation, and mitochondrial proliferation. mtDNA content is reduced in liver. Disease progression is variable, with life expectancy from onset of symptoms ranging from 3 months to 12 years 56).
Mutations in C10orf2 have been associated with variable phenotypes, including infantile-onset spinocerebellar ataxia 57), autosomal dominant progressive external ophthalmoplegia 58) and hepatocerebral mitochondrial DNA depletion syndrome 59). Mutations in C10orf2 are a rare cause of early-onset hepatocerebral mitochondrial DNA depletion syndrome that has been reported in 5 children from 2 unrelated families. Affected individuals typically present in the neonatal or infantile period with lactic acidosis, hepatomegaly, hypotonia, and psychomotor delay. The neurologic involvement progresses to include hyporeflexia, muscular atrophy, ophthalmoplegia, nystagmus, athetosis, ataxia, epilepsy, sensory neuropathy, sensorineural hearing impairment, psychomotor regression. Liver involvement includes cholestasis, increased transaminases, and coagulopathy. Affected infants may also have feeding difficulties and growth retardation. Lactate is increased in plasma and CSF. Neuroimaging can show cerebellar cortical atrophy. Electron transport chain (ETC) activity assays show reduced activities of complexes I, III, and IV. mtDNA content is severely reduced in liver tissue. Prognosis is poor with 3 of the reported affected children dying between 2 and 3 years of age 60).
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) disease
Mutations in TYMP have been reported in about 70 individuals with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) disease 61). Affected individuals usually present clinical manifestations between the first and fifth decades with the majority starting with symptoms before age 20 years. All affected individuals develop weight loss and progressive gastrointestinal dysmotility manifesting as early satiety, nausea, dysphagia, gastroesophageal reflux, postprandial emesis, episodic abdominal pain and distention, and diarrhea. In addition, all affected individuals have peripheral demyelinating motor and sensory neuropathy that may be accompanied by axonal neuropathy in some cases. The neuropathy typically presents with distal weakness and paresthesias occurring in a symmetric stocking-glove distribution. Ptosis and ophthalmoplegia are common findings. Intellectual disability occurs in some individuals. Other variable manifestations include hepatic cirrhosis with increased liver enzymes and macrovesicular steatosis, anemia, sensorineural hearing loss, short stature, autonomic nervous system dysfunction (usually orthostatic hypotension), bladder dysfunction, ventricular hypertrophy, and diverticulosis 62).
Affected individuals can have elevated CSF protein and plasma lactate. Thymidine and deoxyuridine are increased in plasma. In affected individuals, thymidine phosphorylase enzyme activity in leukocytes is usually less than 10 % of the control mean 63). EMG and nerve conduction velocity show decreased motor and sensory nerve conduction velocities, and myopathic changes. Neuroimaging typically demonstrates diffuse white matter abnormalities (leukoencephalopathy) 64). mtDNA depletion, mitochondrial proliferation, and smooth cell atrophy are observed in the external layer of the muscularis propria in the stomach and in the small intestine. Skeletal muscle generally shows histologic abnormalities of a mitochondrial myopathy including ragged-red fibers and defects in single or multiple ETC complexes with the most common defect in complex IV. However, mitochondrial neurogastrointestinal encephalomyopathy has been reported without skeletal muscle involvement at the morphological, enzymatic, or mtDNA content level 65).
Mitochondrial neurogastrointestinal encephalomyopathy is a progressive disease with mean age of death is approximately 40 years (ranging from 25–60 years) 66).
Mitochondrial DNA depletion syndrome diagnosis
Mitochondrial DNA depletion syndrome are multi-organ disorders; therefore, affected individuals should have a comprehensive evaluation to assess the degree of involvement of different systems, including the neuromuscular, hepatic, gastrointestinal, cardiac, and renal systems.
Almost all affected individuals with mitochondrial DNA depletion syndrome show neuromuscular manifestations; therefore, a neurology consultation with comprehensive neurologic examination and developmental/cognitive assessment are mandatory. The following diagnostic modalities can be used to assess the degree of neurological involvement: neuroimaging (mainly brain magnetic resonance imaging) to establish the degree of central nervous system, nerve conduction velocity to establish the degree of the peripheral nervous system involvement, EMG to assess myopathy, and electroencecephalography if seizures are suspected. A thorough ophthalmologic and hearing evaluation is also required.
The degree of liver involvement in the hepatocerebral forms of mitochondrial DNA depletion syndrome can be assessed by liver function tests, including liver transaminases, GGT, albumin, fasting blood glucose, ammonia, and coagulation profile; ultrasound examination to assess liver size and texture, and for the presence of masses; alpha fetoprotein (AFP) to screen for hepatocellular carcinoma; and hepatology/liver transplantation consultation.
Gastrointestinal evaluation in mitochondrial neurogastrointestinal encephalomyopathy disease may depend on the symptoms and can include the following: gastrointestinal consultation, abdominal imaging (X-ray and computed tomography), upper gastrointestinal contrast radiography, esophagogastroduodenoscopy, sigmoidoscopy, liquid phase scintigraphy, and antroduodenal manometry. These studies may show hypoperistalsis, gastroparesis, dilated duodenum, and diverticulosis. Small bowel manometry shows reduced amplitude of contractions 67).
Echocardiogram and electrocardiogram are needed to determine cardiac involvement. Pulmonary function tests and assessment of blood gases are needed for patients with myopathy to assess for respiratory insufficiency. Nutritional evaluation and swallowing assessment are needed in those with feeding difficulty and growth retardation. Urine analysis, urine electrolytes, and urine amino acids can be performed to assess renal tubulopathy.
Mitochondrial DNA depletion syndrome treatment
Although mitochondrial DNA depletion syndrome are severe disorders with poor prognosis in the majority of affected individuals, no curative therapy is available for any of these disorders.
Management of mitochondrial DNA depletion syndrome should involve a multidisciplinary team, including different specialists and aims to provide supportive care and symptomatic treatment for complications associated with these disorders. Other treatment options for some mitochondrial DNA depletion syndrome include dietary modulation, cofactor supplementation, liver transplantation, and stem cell transplantation.
Symptomatic management for mitochondrial DNA depletion syndrome
Seizures are common features in mitochondrial DNA depletion syndrome with neurological involvement. Seizure control with antiepileptic medications is the goal of treatment; however, refractory epilepsy may be very difficult to control. The use of high-dose anticonvulsants and/or treatment with more than 1 medication often becomes necessary to control refractory seizures. It is very important to avoid valproic acid (Depakene®) and sodium divalproate (divalproex) (Depakote®) in treating seizures in mitochondrial DNA depletion syndrome, particularly POLG-related disorders, because of the risk of precipitating and/or accelerating liver disease 68).
Physical therapy can help maintain muscle function and prevent joint contractures. Feeding difficulties and failure to thrive may require nutritional support by experienced dietitian, occupational therapy to improve oromotor functions, and the use of a nasogastric tube or gastrostomy tube feedings.
Respiratory insufficiency can benefit from chest physiotherapy, aggressive antibiotic treatment of chest infections, and artificial ventilation that could include assisted nasal ventilation or intubation, and the use of a tracheostomy and ventilator.
Other treatment options include bracing to treat scoliosis or kyphosis, surgery for ptosis, and cochlear implantation for sensorineural hearing loss.
Nutritional modulation in mitochondrial DNA depletion syndrome
Formulas with an enriched medium-chain triglyceride content may provide better nutritional support for infants with cholestasis than formulas with predominantly long-chain triglycerides 69).
Prevention of hypoglycemia requires avoidance of fasting by frequent or continuous feeding. In addition, uncooked cornstarch may reduce symptomatic hypoglycemia in individuals with DGUOK and MPV17-related hepatocerebral mitochondrial DNA depletion syndrome 70). Furthermore, cornstarch use may slow the progression of the liver disease in MPV17-related hepatocerebral mitochondrial DNA depletion syndrome 71).
Cofactor use in mitochondrial DNA depletion syndrome
Succinate and ubiquinone were reported to slow the progression of liver impairment in MPV17-related mitochondrial DNA depletion syndrome 72).
Elevated CSF inflammatory cytokines and blocking folate receptor autoantibodies associated with reduced CSF folate were reported in a child with Alpers-Huttenlocher syndrome. Treatment with oral folinic acid (leucovorine) resulted in improvement of CSF folate level and seizure frequency, and communicative abilities improved 73). Therefore, CSF folate may be deficient in disorders that lead to mtDNA depletion. It has been suggested that testing for CSF folate deficiency with treatment offered to those with deficiency can be one option; the other option can be empiric therapy with folinic acid 74).
Levocarnitine, creatine monohydrate, coenzyme Q10, B vitamins, and antioxidants, such as alpha lipoic acid, vitamin E, and vitamin C, have been used as mitochondrial supplements. These cofactors have been used in mitochondrial DNA depletion syndrome; however, there is very limited evidence for their effectiveness 75).
More recently, enteral administration of sodium pyruvate to a child with myopathic mitochondrial DNA depletion syndrome has been reported to improve muscle strength and quality of life score using used the Newcastle Pediatric Mitochondrial Disease Scale. No significant change of the blood lactate level or lactate-to-pyruvate ratio were noticed 76). Further evaluation is need before reaching conclusions about the effectiveness of such therapy.
Studying myotube cells of individuals with mitochondrial DNA depletion syndrome have demonstrated that the application of variable combinations of deoxynucleoside monophosphates in different types of mitochondrial DNA depletion syndrome result in near normalization of mtDNA content in many cases. Therefore, the use of deoxynucleoside monophosphate combinations may be a possible therapeutic approach for individuals with mitochondrial DNA depletion syndrome 77). Further clinical investigation is needed to investigate this approach.
Liver transplantation in mitochondrial DNA depletion syndrome
Although liver transplantation remains the only treatment option for liver failure in hepatocerebral mitochondrial DNA depletion syndrome, liver transplantation in mitochondrial hepatopathy is controversial, largely because of the multi-organ involvement.
Liver transplantation has been performed in about a third of affected individuals with MPV17-related mitochondrial DNA depletion syndrome; the outcome has not been satisfactory, with half of the transplanted children dying in the post-transplantation period because of multi-organ failure and/or sepsis 78).
For children with multi-organ DGUOK-related mitochondrial DNA depletion syndrome, liver transplantation provides no survival benefit. However, several children with isolated hepatic disease have had excellent 10-year survival with liver transplantation and, thus, it is a potential therapeutic option. However, this option warrants careful discussion with parents because at least 1 child with isolated liver disease developed neurologic features after liver transplantation 79).
Liver transplantation is not advised in children with Alpers-Huttenlocher syndrome because transplanting the liver does not alter the rapid progression of the neurological complications 80). However, liver transplantation in adults who have an acceptable quality of life may be of benefit. Two affected individuals with POLG-related disorders were reported to survive after liver transplantation 81).
Thymidine reduction in mitochondrial neurogastrointestinal encephalomyopathy disease
In mitochondrial neurogastrointestinal encephalomyopathy, a correlation between plasma thymidine levels and the severity of the phenotype has been observed 82). Therefore, it has been proposed that the reduction in circulating thymidine levels can result in disease improvement. Peritoneal dialysis has been used to reduce the thymidine levels leading to an improvement of the symptoms in affected individuals with mitochondrial neurogastrointestinal encephalomyopathy disease 83). Enzyme replacement therapy via infusion of platelets from healthy donors to individuals with mitochondrial neurogastrointestinal encephalomyopathy resulted in reduction of circulating thymidine and partially restored thymidine phosphorylase activity 84).
Allogeneic hematopoietic stem cell transplantation (HSCT) offers the possibility of sustained correction of enzyme deficiency and has become an established treatment for many different storage diseases. More than 10 individuals with mitochondrial neurogastrointestinal encephalomyopathy disease have so far been treated with allogeneic hematopoietic stem cell transplantation 85). Allogeneic hematopoietic stem cell transplantation has been shown to restore thymidine phosphorylase activity, lowering thymidine levels and improving the gastrointestinal dysmotility [132–135]. However, neurological assessments remained unchanged 86). Although hematopoietic stem cell transplantation corrects biochemical abnormalities and improves gastrointestinal symptoms, the procedure can be risky in patients already in poor medical condition, as are many mitochondrial neurogastrointestinal encephalomyopathy patients and several affected patients were reported to die in the post-transplantation period 87). As transplant-related morbidity and mortality increase with the progression of the disease and the number of associated comorbidities, it has been suggested that individuals with mitochondrial neurogastrointestinal encephalomyopathy should be referred to hematopoietic stem cell transplantation when they are still relatively healthy in order to minimize the complications of the procedure 88).
References [ + ]




{kind=link}