
Contents
- What is leukodystrophy
- What is myelin?
- Leukodystrophy types
- Adult-onset autosomal dominant leukodystrophy
- Aicardi-Goutieres syndrome
- Alexander disease
- CADASIL
- Canavan disease
- CARASIL
- Cerebrotendinous xanthomatosis
- Childhood ataxia with cerebral hypomyelination
- Fabry disease
- Fucosidosis
- GM1 gangliosidosis
- L-2-hydroxyglutaric aciduria
- Megalencephalic leukoencephalopathy with subcortical cysts
- Multiple sulfatase deficiency
- Pelizaeus-Merzbacher disease
- Pol III-Related Leukodystrophies
- Refsum disease
- Salla disease
- Sjögren-Larsson syndrome
- X-linked adrenoleukodystrophy
- Zellweger syndrome spectrum disorders
- Krabbe leukodystrophy
- Metachromatic leukodystrophy
- Leukodystrophy prognosis
- Leukodystrophy life expectancy
- Leukodystrophy symptoms
- Leukodystrophy treatment
What is leukodystrophy
Leukodystrophies make up a group of rare heritable myelin disorders affecting the white matter of the central nervous system (the brain and spinal cord) with or without peripheral nervous system (the rest of the neurons in the body) myelin involvement 1). The word leukodystrophy comes from the Greek words leuko (meaning white), dys (meaning ill), and trophy (meaning growth). Adding these pieces together, the word leukodystrophy describes a disease that affects the growth or maintenance of the white matter (myelin). Leukodystrophies disrupt the growth or maintenance of the myelin sheath, which insulates nerve cells. Leukodystrophies are progressive, meaning that they tend to worsen throughout the life of the patient. Involvement of the white matter tracts almost universally leads to motor involvement that manifests as hypotonia in early childhood and progresses to spasticity over time. This may lead to variable motor impairment, from mild spastic diplegia to severe spastic quadriplegia that limits purposeful movement. In addition, motor dysfunction is likely to significantly impair vital functions including swallowing, chewing, and (in some cases) respiration. Other findings that vary by disorder include extrapyramidal movement disorders (e.g., dystonia and/or dyskinesias), ataxia, seizures, and delay in cognitive development or change in cognitive function over time.
All leukodystrophies are a result of problems with the growth or maintenance of the myelin sheath. There are many genes that are important in this process. For example, some genes are involved with the synthesis of the proteins needed for the myelin, while others are required for the proper transport of these proteins to their final location in the myelin sheath that covers the axons. Defects in any of the genes (called a mutation) may lead to a leukodystrophy; however, the symptoms of the individual leukodystrophies may vary because of the differences in their genetic causes.
Leukodystrophies are mostly inherited disorders, meaning that they pass from parent to child. Leukodystrophy may be inherited in an autosomal dominant manner, an autosomal recessive manner, or an X-linked recessive manner; other inheritance patterns may be identified as more genetic causes of leukodystrophy are discovered and depends on the type of leukodystrophy.
There are some leukodystrophies that do not appear to be inherited, but rather arise spontaneously. They are still caused by a mutation in a particular gene, but it means that the mutation was not inherited. In this case, the birth of one child with the disease does not necessarily increase the likelihood of a sibling having the disease.
Genetic counseling regarding risk to family members depends on accurate diagnosis, determination of the mode of inheritance in each family, and results of molecular genetic testing. Prenatal testing for pregnancies at increased risk is possible for some types of leukodystrophy if the pathogenic variant(s) in the family are known. Many leukodystrophies are still without an identified genetic cause; once a genetic cause is identified, other inheritance patterns may emerge.
Genetic counseling may help you understand the risks of passing leukodystrophy on to any children you have.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Treatment is symptomatic and ideally occurs in a multidisciplinary setting by specialists experienced in the care of persons with a leukodystrophy. Pharmacologic agents are used to manage muscle tone and block neuronal signaling to muscle (chemodenervation). Intensive physical therapy is used to improve mobility and function. Pharmacologic treatment of dystonia and dyskinesias may result in significant functional improvement. Treatment of ataxia, seizures, and cognitive issues is provided in the usual manner, depending on the needs of the individual.
Prevention of primary manifestations: In a few leukodystrophies primary disease manifestations can be prevented by hematopoietic stem cell transplantation (HSCT) or bone marrow transplantation (BMT) early in the disease course.
Surveillance: Routine assessment of growth and nutritional status; physical examination and/or serial x-rays of the hips and spine to monitor for orthopedic complications; and routine history re signs and symptoms of seizures.
Agents/circumstances to avoid: Mild head injuries and infection as these may exacerbate disease manifestations.
Evaluation of relatives at risk: When primary prevention of a leukodystrophy is possible (e.g., by HSCT or BMT), it is appropriate to offer testing to asymptomatic at risk relatives who would benefit from early diagnosis and consideration of early treatment.
The leukodystrophies do share some common features with multiple sclerosis (MS). Like the leukodystrophies, MS is caused by the loss of myelin from the axons; however, the cause is different. Whereas leukodystrophies are generally caused by a defect in one of the genes involved with the growth or maintenance of the myelin, MS is thought to be caused by an attack on the myelin by the body’s own immune system.
What is myelin?
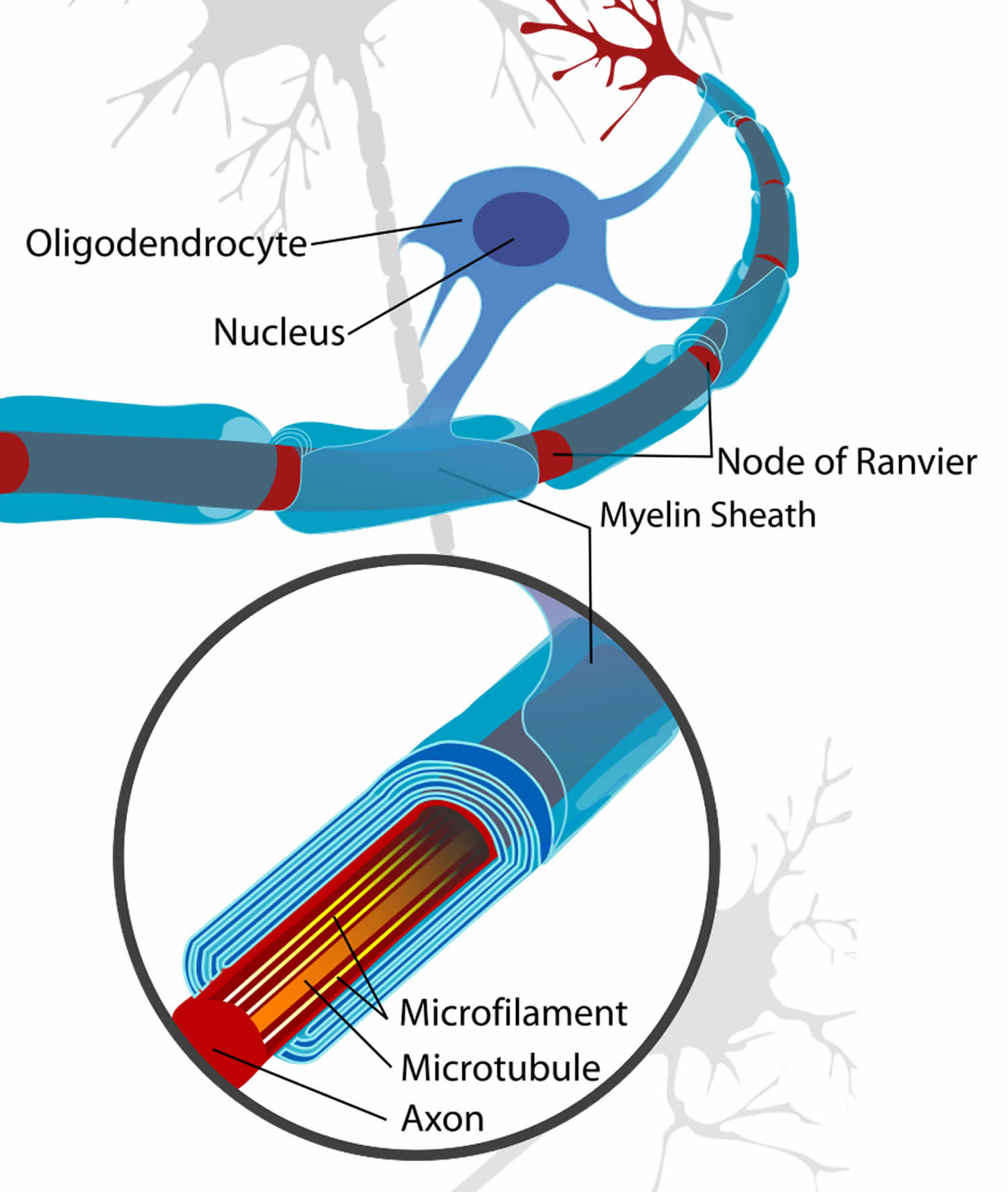
Myelin is a lipid-rich (fatty) substance, sometimes referred to as “white matter” because of its white, fatty appearance, protects and insulates the axons. Myelin consists of a protective sheath of many different molecules that include both lipids (fatty molecules) and proteins. Myelin is an electrical insulator that insulates nerve cell axons to increase the speed at which information (encoded as an electrical signal) travels from one nerve cell body to another (as in the central nervous system) or from a nerve cell body to a muscle (as in the peripheral nervous system). The myelinated axon can be likened to an electrical wire (the axon) with insulating material (myelin) around it. However, unlike the plastic covering on an electrical wire, myelin does not form a single long sheath over the entire length of the axon. Rather, each myelin sheath insulates the axon over a single section and in general, each axon comprises multiple long myelinated sections separated from each other by short gaps called the nodes of Ranvier. Nodes of Ranvier are the short (~1 micron) unmyelinated regions of the axon between adjacent long (~0.2 mm – >1 mm) myelinated internodes 2). Each myelin sheath is formed by the concentric wrapping of an oligodendrocyte or Schwann cell process around the axon. Each myelin-generating cell (oligodendrocyte in the CNS or Schwann cell in the peripheral nervous system) furnishes myelin for only one segment of any given axon. The periodic interruptions where short portions of the axon are left uncovered by myelin, the nodes of Ranvier, are critical to the functioning of myelin.
With the protective myelin coat, neurons can transmit signals at speeds up to 60 meters per second. When the coat is damaged, the maximum speed can decrease by ten-fold or more, since some of the signal is lost during transmission. This decrease in speed of signal transmission leads to significant disruption in the proper functioning of the nervous system.
In myelinated axons, the excitable axonal membrane is exposed to the extracellular space only at the nodes of Ranvier; this is the location of sodium channels 3). When the membrane at the node of Ranvier is excited, the local circuit generated cannot flow through the high-resistance sheath and, therefore, flows out through and depolarizes the membrane at the next node, which might be 1 mm or farther away (Figure 3). The low capacitance of the myelin sheath means that little energy is required to depolarize the remaining membrane between the nodes of Ranvier, which results in local circuit spreading at an increased speed. Active excitation of the axonal membrane jumps from node to node; this form of impulse propagation is called saltatory conduction (Latin saltare, “to jump”). This saltatory conduction whereby the action potential “jumps” from one node of Ranvier, over a long myelinated stretch of the axon called the internode, before ‘recharging’ at the next node of Ranvier, and so on, until it reaches the axon terminal. Once it reaches the axon terminal, this electrical signal provokes the release of a chemical message or neurotransmitter that binds to receptors on the adjacent post-synaptic cell (e.g. nerve cell in the CNS or muscle cell in the peripheral nervous system) at specialized regions called synapses.
Furthermore, such movement of the wave of depolarization is much more rapid in myelinated nerve fibers than in unmyelinated fibers, because only the nodes of Ranvier are excited during conduction in myelinated fibers, Na+ (sodium) flux into the nerve is much less than in unmyelinated fibers, where the entire membrane is involved. An example of the advantage of myelination is obtained by comparison of two different nerve fibers, both of which conduct at 25 m/sec at 20°C. The 500-mm diameter unmyelinated giant axon of the squid requires 5,000 times as much energy and occupies about 1,500 times as much space as the 12-mm diameter myelinated nerve in the frog.
In another word, myelin speeds the transmission of electrical impulses called action potentials along myelinated axons by insulating the axon and reducing axonal membrane capacitance. Conduction velocity in myelinated fibers is proportional to the diameter, while in unmyelinated fibers it is proportional to the square root of the diameter 4). Thus, differences in energy and space requirements between the two types of fiber are exaggerated at higher conduction velocities. If nerves were not myelinated and equivalent conduction velocities were maintained, the human spinal cord would need to be as large as a good-sized tree trunk. Myelin, then, facilitates conduction while conserving space and energy 5).
This “insulating” role for myelin is essential for normal motor function (i.e. movement such as walking), sensory function (e.g. hearing, seeing or feeling the sensation of pain) and cognition (e.g. acquiring and recalling knowledge), as demonstrated by the consequences of disorders that affect it, such as the genetically determined leukodystrophies 6), the acquired inflammatory demyelinating disorder, multiple sclerosis 7) and the inflammatory demyelinating peripheral neuropathies 8). Due to its high prevalence, multiple sclerosis, which specifically affects the central nervous system (brain, spinal cord and optic nerve), is the best known disorder of myelin.
Figure 1. Neuron with myelin sheath
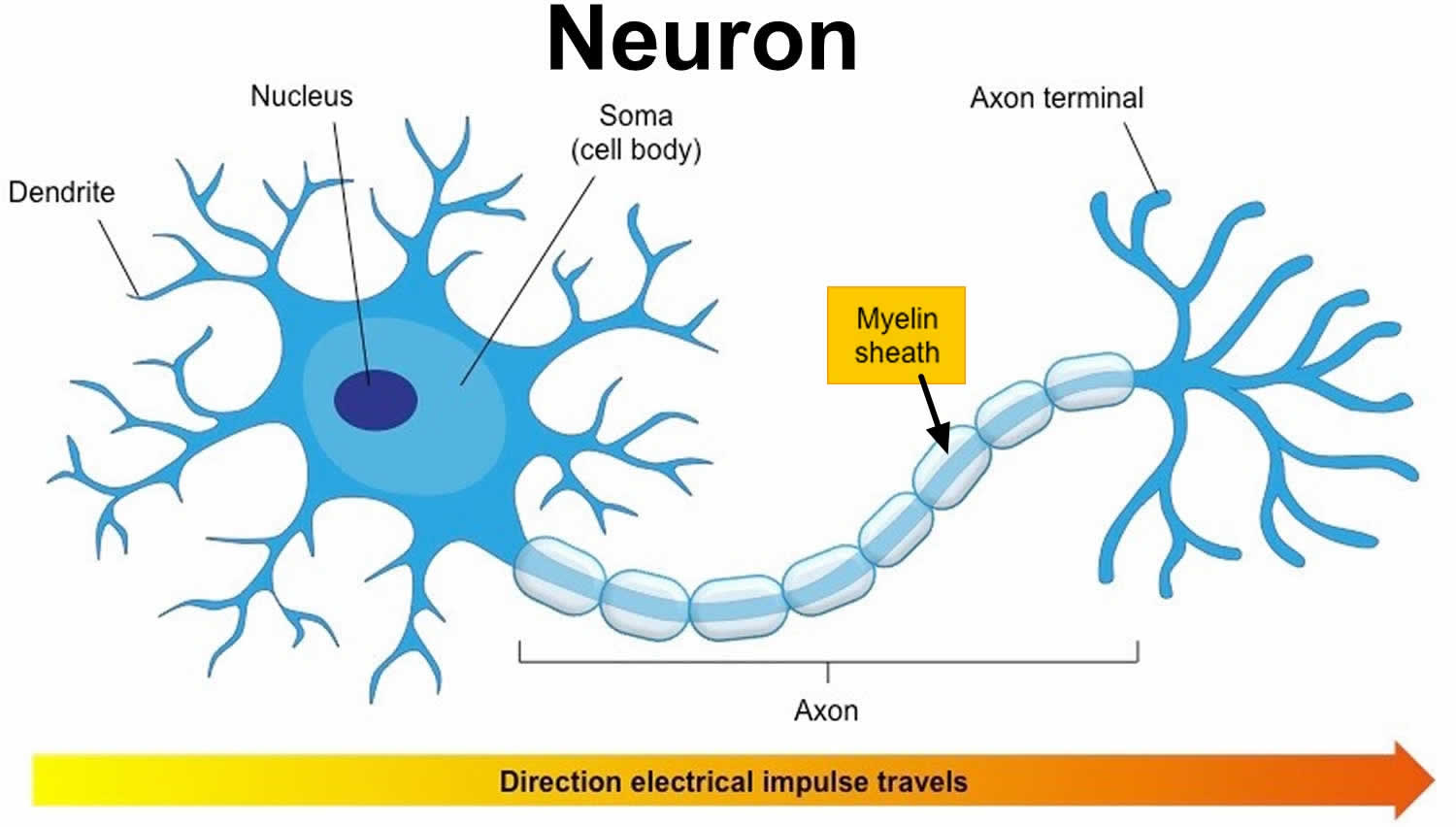

Figure 2. How electrical impulses travel down a neuron (myelinated and unmyelinated)
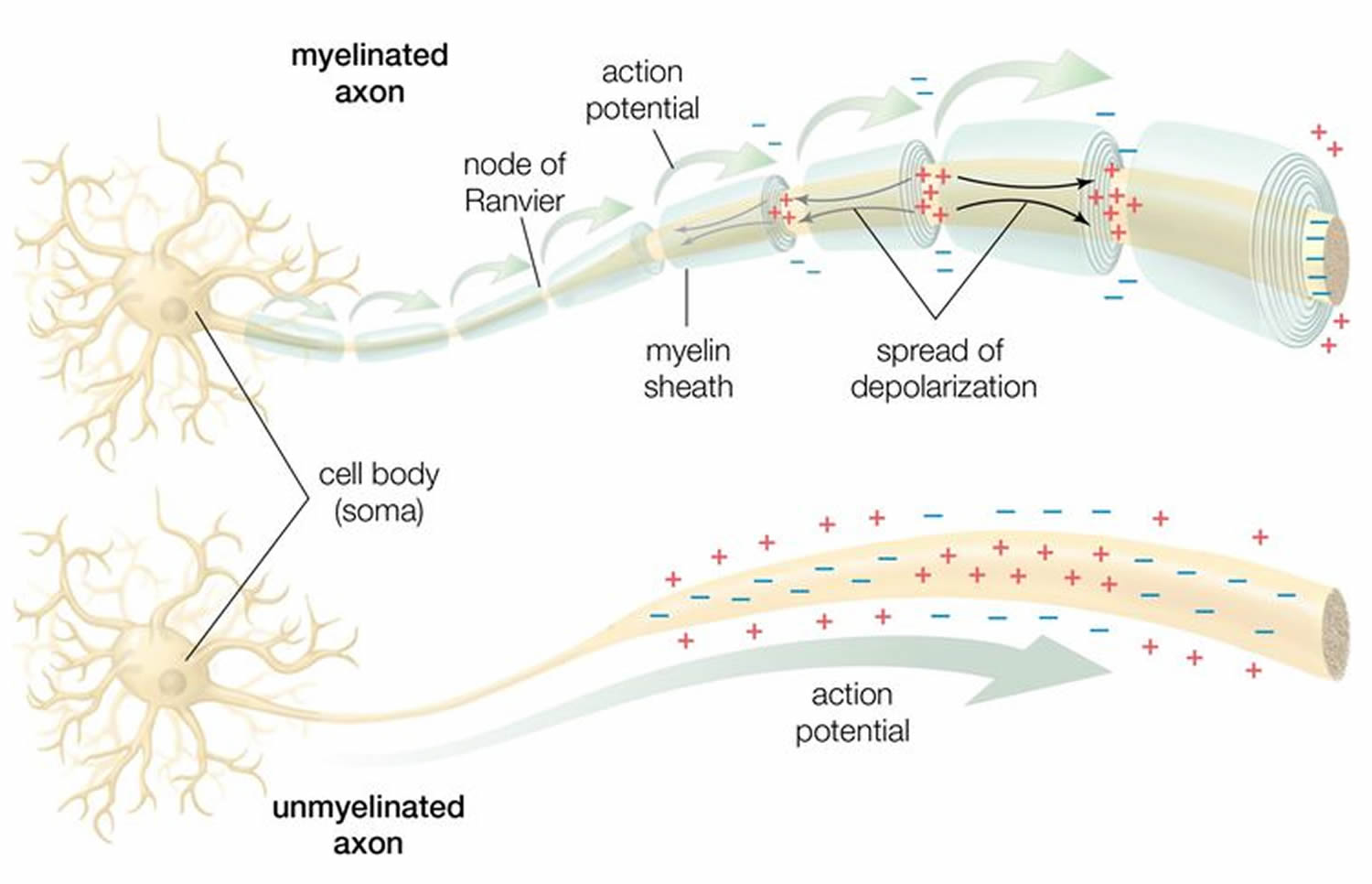
Figure 3. Formation of myelin sheath in the central nervous system (CNS)
Leukodystrophy types
Known types of leukodystrophy and leukoencephalopathy (in alphabetical order) 9)
- 18q Syndrome with Deficiency of Myelin Basic Protein
- Acute Disseminated Encephalomyeolitis (ADEM)
- Acute Disseminated Leukoencephalitis
- Acute Hemorrhagic Leukoencephalopathy
- Adrenoleukodystrophy (ALD) – See X-linked Adrenoleukodystrophy
- Adrenomyeloneuropathy (AMN)
- Adult Onset Autosomal Dominant Leukodystrophy (ADLD)
- Adult Polyglucosan Body Disease
- Aicardi-Goutieres Syndrome
- Alexander Disease
- Autosomal Dominant Diffuse Leukoencephalopathy with Neuroaxonal Spheroids (HDLS)
- Autosomal Dominant Late-Onset Leukoencephalopathy
- Canavan Disease
- Childhood Ataxia with Diffuse CNS Hypomyelination (CACH or Vanishing White Matter Disease)
- Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL)
- Cerebroretinal Micro-Angiography with Calcifications and Cysts
- Cerebrotendinous Xanthomatosis (CTX)
- Childhood Ataxia with Central Nervous System Hypomyelination (CACH) – See Vanishing White Matter Disease
- Craniometaphysical Dysplasia with Leukoencephalopathy
- Cystic Leukoencephalopathy (RNASET2 related)
- Elongation of Very Long-Chain Fatty Acids-4 (ELOVL4; Pseudo-Sjogren-Larsson)
- Extensive Cerebral White Matter Abnormality without Clinical Symptoms
- Familial Adult-Onset Leukodystrophy Manifesting as Cerebellar Ataxia and Dementia
- Familial Leukodystrophy with Adult Onset Dementia and Abnormal Glycolipid Storage
- Fatty Acid 2-Hydroxylase Deficiency
- Fucosidosis
- Fukuyama Congential Muscular Dystrophy
- Galactosialidosis
- Globoid Cell Leukodystrophy (Krabbe Disease)
- GM1 Gangliosidosis
- GM2 Gangliosidosis (Tay-Sachs Disease)
- Hereditary Adult Onset Leukodystrophy Simulating Chronic Progressive Multiple Sclerosis
- Herditary Diffuse Leukoencephalopathy with Axonal Spheroids (HDLS)
- Hypomyelination with Atrophy of the Basal Ganglia and Cerebellum (H-ABC)
- Hypomyelination, Hypogonadotropic, Hypogonadism and Hypodontia (4H Syndrome)
- Leukoencephalopathy with Brain Stem and Spinal Cord Involvement and Lactate Elevation (LBSL)
- Lipomembranous Osteodysplasia with Leukodystrophy (Nasu Disease)
- Metachromatic Leukodystrophy (MLD)
- Megalencephalic Leukodystrophy with subcortical Cysts (MLC)
- Neuroaxonal Leukoencephalopathy with axonal spheroids (Hereditary diffuse leukoencephalopathy with spheroids – HDLS)
- Neonatal Adrenoleukodystrophy (NALD)
- Oculodetatoldigital Dysplasia with Cerebral White Matter Abnormalities
- Orthochromatic Leukodystrophy with Pigmented Glia
- Ovarioleukodystrophy Syndrome
- Pelizaeus Merzbacher Disease (X-linked spastic paraplegia)
- Refsum Disease
- Sjogren-Larsson Syndrome
- Sudanophilic Leukodystrophy – See Adrenoleukodystrophy – ALD
- Van der Knaap Syndrome (Vacuolating Leukodystrophy with Subcortical Cysts or MLC)
- Vanishing White Matter Disease (VWM) or Childhood Ataxia with Diffuse Central Nervous System Hypomyelination (CACH)
- X-linked Adrenoleukodystrophy (X-ALD)
- Zellweger Spectrum: Zellweger Syndrome, Neonatal Adrenoleukodystrophy, and Infantile Refsum Disease
Table 1. Leukodystrophies Meeting Strict Diagnostic Criteria
| Name of Disorder | Mode of Inheritance | Gene 1 | Biochemical Testing / Other |
|---|---|---|---|
| 18q deletion syndrome | Most often de novo deletion; may be inherited | Chromosome analysis for 18q microdeletion involving MBP | |
| Adult polyglucosan body disease (APBD) | AR | GBE1 | Histopathologic examination of muscle, nerve, axillary skin: pathologic polyglucosan accumulation |
| Aicardi-Goutières syndrome (AGS) | Usually AR; may be AD | TREX1 RNASEH2A RNASEH2B RNASEH2C SAMHD1 ADAR |
CSF analysis: lymphocytosis, ↑ interferon-α, ↑ pterins |
| Alexander disease | AD | GFAP | |
| AD adult-onset leukodystrophy (ADLD) | AD | LMNB1 | |
| Cerebroretinal microangiopathy w/calcifications & cysts (CRMCC) 2 | Likely AR | Clinical & neuroradiologic features | |
| Canavan disease | AR | ASPA | In urine, plasma, CSF, & amniotic fluid: ↑ N-acetylaspartic acid in urine; In skin fibroblasts: deficient aspartoacylase enzyme activity |
| Cerebrotendinous xanthomatosis (CTX) | AR | CYP27A1 | In plasma & CSF: ↑ cholestanol concentration, ↓ chenodeoxycholic acid; In bile, urine, plasma: ↑ concentration bile alcohols & glyconjugates; In fibroblasts, liver, leukocytes: ↓ sterol 27-hydroxylase activity |
| Childhood ataxia w/CNS hypomyelination / vanishing white matter (CACH/VWM) | AR | EIF2B1-5 | |
| Free sialic acid storage disorders 3 | AR | SLC17A5 | In urine, fibroblast, lysosomes: ↑ free sialic acid |
| Fucosidosis | AR | FUCA1 | On urinary oligosaccharide assay: ↑ fucose-containing glycoconjugates; In leukocytes or fibroblasts: deficient α-fucosidase activity |
| Hypomyelination w/atrophy of the basal ganglia & cerebellum (H-ABC) | Likely AD | TUBB4A | Clinical & neuroradiologic features |
| Hypomyelination and congenital cataract (HCC) | AR | FAM126A | |
| Krabbe disease | AR | GALC See footnote 4 |
In leukocytes or fibroblasts: deficient galactocerebrosidase activity |
| L-2-hydroxyglutaric aciduria | AR | L2HGDH | In plasma, urine, CSF: ↑ concentration of L-2-hydroxyglutaric acid (and lysine) |
| Leukoencephalopathy w/brain stem & spinal cord involvement & lactate elevation (LBSL) | AR | DARS2 | |
| Leukoencephalopathy w/thalamus and brain stem involvement & lactate elevation (LTBL) | AR | EARS2 | |
| Megalencephalic leukodystrophy w/subcortical cysts (MLC) | AR | MLC1 HEPACAM (MLC2) |
|
| Metachromatic leukodystrophy (MLD) | AR | ARSA | In leukocytes, fibroblasts: ↓ arylsulfatase A activity; In urine: ↑ sulfatides |
| PSAP-related MLD 5 | PSAP | In leukocytes, fibroblasts: normal arylsulfatase A activity; In urine: ↑ sulfatides |
|
| Multiple sulfatase deficiency (MSD) | SUMF1 | ↓ activity of other sulfatases; In urine: ↑ mucopolisaccharides, ↑ urinary sulfatides |
|
| Hereditary diffuse leukoencephalopathy w/spheroids (HDLS) 6 | AD | CSF1R | |
| Oculodentodigital dysplasia (ODDD) | Usually AD; may be AR | GJA1 | |
| Pelizaeus-Merzbacher disease (PMD) | XL | PLP1 | |
| Pelizaeus-Merzbacher-like disease 1 (PMLD1) | AR | GJC2 | |
| Zellweger spectrum disorder (PBD, ZSD) 7 | AR | PEX genes | Plasma VLCFA, phytanic & pristanic acid, plasma & urine concentration of pipecolic acid & bile acids aid to distinguish different forms of peroxisomal disorders |
| Pol III-related leukodystrophies 8 | AR | >POLR3A POLR3B |
|
| RNAse T2-deficient leukoencephalopathy | AR | RNASET2 | |
| Single-enzyme deficiencies of peroxisomal fatty acid beta oxidation 9 | AR | Dibifunctional protein deficiency: HSD17B4 | Plasma VLCFA, phytanic & pristanic acid, plasma & urine concentration of pipecolic acid & bile acids aid to distinguish different forms of peroxisomal disorders |
| Peroxisomal acyl-CoA-oxidase deficiency: ACOX1 | |||
| SCPx deficiency: SCP2 | |||
| Sjögren-Larsson syndrome | AR | ALDH3A2 | In urine: abnormal metabolites of leukotriene B4; In cultured skin fibroblasts, leukocytes: deficiency of fatty aldehyde dehydrogenase activity (FALDH) and/or of fatty alcohol:NAD oxidoreductase (FAO) |
| SOX10-associated disorders | AD | SOX10 | |
| X-linked adrenoleukodystrophy (X-ALD) | XL | ABCD1 | On plasma VLCFA assay: C26:0, ↑ ratio of C24:0 to C22:0, ↑ ratio of C26:0 to C22:0 |
Footnotes:
Disorders listed in alphabetic order.
- Genetic testing is available for many of these genes.
- This disorder now appears to be distinct from Coats plus caused by pathogenic variants in CTC1, encoding conserved telomere maintenance component 1.
- Includes Salla disease; infantile sialic acid storage disease, intermediate form.
- Defects in PSAP causing a deficiency in the activator protein of SapA-d essential for the action of GALC have been reported.
- Pathogenic variants in PSAP result in deficiency in SapB-d, an activator protein essential for ARSA activity.
- Also known as adult-onset leukodystrophy w/ neuroaxonal spheroids & pigmented glia; may include hereditary diffuse; pigmentary type of orthochromatic leukodystrophy w/pigmented glia (POLD).
- Includes neonatal adrenoleukodystrophy; infantile Refsum disease.
- Includes hypomyelination, hypodontia, hypogonadotropic hypogonadism (4H syndrome); ataxia, delayed dentition, and hypomyelination (ADDH); tremor-ataxia with central hypomyelination (TACH); leukodystrophy with oligodontia (LO); and hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum (HCAHC).
- Includes D-bifunctional protein (DBP) deficiency; sterol carrier protein-2 (SCPx) deficiency; peroxisomal acyl-CoA-oxidase deficiency
Abbreviations: AD = autosomal dominant; AR = autosomal recessive; XL = X-linked; VLCFA = very long-chain fatty acid
[Source 10) ]Adult-onset autosomal dominant leukodystrophy
Adult-onset autosomal dominant leukodystrophy results from tandem duplication of the LMNB1 gene, which encodes the nuclear lamina protein lamin B1. Symptoms begin in the fourth to fifth decade with autonomic dysfunction including bowel and bladder dysfunction and orthostatic hypotension with lightheadedness. This is followed by slowly progressive motor and balance difficulties. The MRI of the brain shows extensive white matter involvement with relative sparing of the periventricular white matter. The spinal cord develops atrophy which may precede the motor difficulties.
Aicardi-Goutieres syndrome
Aicardi-Goutieres syndrome is an autosomal recessive condition, presenting with an early encephalopathy followed by stabilization of neurologic symptoms. At least six different genes have been described. Neuroimaging reveals leukoencephalopathy with calcifications and cerebral atrophy. Cerebrospinal fluid analysis reveals chronic lymphocytosis (elevated white blood cell count), elevated INF-a, and neopterin.
Alexander disease
Alexander disease is a rare, progressive, leukodystrophy that usually becomes apparent during infancy or early childhood but juvenile and adult onset forms have also been reported. Alexander disease is characterized by degenerative changes of the white matter of the brain caused by a lack of normal amounts of myelin. The disorder is also associated with the formation of abnormal, fibrous deposits known as “Rosenthal fibers” in the astrocytic processes around small blood vessels and astrocytic cell bodies in certain regions of the brain and spinal cord. The disease is caused by a dominant gain of function mutation in the glial fibrillary acidic protein (GFAP) (Chromosome 17q21). Treatment for Alexander’s disease is currently symptomatic consisting of anticonvulsants for seizures, orthopedic and pharmacologic management of spasticity, and nutritional support. Strategies for future treatment include decreasing the expression of GFAP.
CADASIL
CADASIL is a rare genetic disorder with dominant inheritance caused by a mutation in the NOTCH3 receptor gene. This condition presents with migraine headaches and multiple strokes in adults, even young adults, often without cardiovascular risk factors. CADASIL often progresses to cause cognitive impairment and dementia. The symptoms of CADASIL result from damage of various small blood vessels, especially those within the brain. The age of onset, severity, specific symptoms and disease progression varies greatly from one person to another, even among members of the same family. CADASIL is an acronym that stands for:
- (C)erebral – relating to the brain
- (A)utosomal (D)ominant – a form of inheritance in which one copy of an abnormal gene is necessary for the development of a disorder
- (A)rteriopathy – disease of the small arteries (blood vessels that carry blood away from the heart)
- (S)ubcortical – relating to a specific area of the deep brain that is involved in higher functioning (e.g., voluntary movements, reasoning, memory)
- (I)nfarcts – tissue loss in the brain caused by lack of oxygen to the brain, which occurs when blood flow in the small arteries is blocked or abnormal
- (L)eukoencephalopathy – destruction of the myelin, that covers and protects nerve fibers in the central nervous system
Canavan disease is a rare inherited neurological disorder characterized by spongy degeneration of the brain and spinal cord (central nervous system). Physical symptoms that appear in early infancy may include progressive mental decline accompanied by the loss of muscle tone, poor head control, an abnormally large head (macrocephaly), and/ or irritability. Physical symptoms appear in early infancy and usually progress rapidly. Canavan disease is caused by an abnormality in the ASPA gene (Chromosome 17p13-ter0) that leads to a deficiency of the enzyme aspartoacylase. Canavan disease is inherited as an autosomal recessive genetic disorder. There are two common mutations among the Ashkenazi Jewish individuals that account for over 97% of the alleles in Jewish patients with Canavan disease.
CARASIL
CARASIL is rare autosomal recessive disorder that is caused by mutations in cerebral small-vessel disease protein HTRA1 that controls the amount of TGF-B1 via cleavage of proTGF-B1b. Individuals with CARASIL are at risk of developing multiple strokes, even if they do not have cardiovascular risk factors. The symptoms of CARASIL result from damage to various small blood vessels, especially those within the brain. Individuals with CARASIL may develop a variety of symptoms relating to white matter involvement or leukoaraiosis (changes in deep white matter in the brain, which are observed on MRI). Such symptoms include an increasing muscle tone (spasticity), pyramidal signs, and pseudo bulbar palsy beginning between 20 and 30 years of age. Pseudo bulbar palsy is a group of neurologic symptoms including difficulties with chewing, swallowing and speech. Eventually, cognitive impairment and dementia may result. About half of cases have a stroke-like episode. The age of onset is 20 to 50 years old. CARASIL is an acronym that stands for:
- (C)erebral – relating to the brain or the cerebellum, which is the part of the brain that controls balance and muscular coordination
- (A)utosomal (R)ecessive – a form of inheritance in which two copies (one from each parent) of an abnormal gene is necessary for the development of a disorder
- (A)rteriopathy – disease of the small arteries (blood vessels that carry blood away from the heart)
- (S)ubcortical – relating to a specific area of the deep brain that is involved in higher functioning (e.g., voluntary movements, reasoning, memory)
- (I)nfarcts – tissue loss in the cerebellum caused by lack of oxygen to the brain, which occurs when blood flow in the small arteries is blocked or abnormal
- (L)eukoencephalopathy – destruction of the myelin, an oily substance that covers and protects nerve fibers in the central nervous system
Cerebrotendinous xanthomatosis
Cerebrotendinous xanthomatosis (CTX) is an autosomal recessive genetic disorder due to mutations in the sterol 27-hydroxylase gene (CYP27A1), resulting in a deficiency of the mitochondrial enzyme sterol 27-hydroxylase. The lack of this enzyme prevents cholesterol from being converted into a bile acid called chenodexoycholic acid. Lipid rich deposits containing cholestanol and cholesterol accumulate in the nerve cells and membranes, and cause damage to the brain, spinal cord, tendons, lens of the eye and arteries. Affected individuals experience cataracts during childhood and benign, fatty tumors (xanthomas) of the tendons during adolescence. The disorder leads to progressive neurologic problems in adulthood such as paralysis, ataxia and dementia. Coronary heart disease is common. More than 300 patients with cerebrotendinous xanthomatosis (CTX) have been reported to date worldwide and about 50 different mutations identified in the CYP27A1 gene. Almost all mutations lead to the absent or inactive form of the sterol 27-hydroxylase. Dietary therapy with the bile acid, chenodeoxycholic acid, does correct many of the symptoms of cerebrotendinous xanthomatosis (CTX); however, early diagnosis of the disorder with early therapy leads to a better clinical outcome. The activity of cholesterol 7 alpha-hydroxylase, the rate limiting enzyme in bile acid synthesis, is normalized by this diet therapy and there is a reduction in the development of xanthomas.
Childhood ataxia with cerebral hypomyelination
Childhood ataxia with cerebral hypomyelination (CACH), also known as vanishing white matter disease (VWMD), is an autosomal recessive leukodystrophy that is characterized by progressive deterioration in motor function and speech during the first five years of life. Clinical symptoms typically begin in the first few years of life, following a normal to mildly delayed early development. Common presenting symptoms include ataxia and seizures. The course is chronic and progressive with episodic decline following fever, head trauma, or periods of fright. Patients usually survive only a few years past the clinical onset, though the course is variable even among patients with mutations in the same eIF2B subunit. In the rare reports of adult-onset vanishing white matter disease (VWMD), the typical presentation consists of cognitive deterioration, pseudo bulbar palsy and progressive spastic paraparesis. An important association between vanishing white matter disease (VWMD) and ovarian failure has been described, termed ‘ovarioleukodystrophy’. Vanishing white matter disease (VWMD) may be one of the more common inherited leukoencephalopathies, though its exact incidence is not yet known.
Vanishing white matter disease (VWMD) is caused by mutations in one of the 5 subunits of eukaryotic initiation factor 2B (eIF2B). eIF2B is a highly conserved, ubiquitously expressed protein that plays an essential role in the initiation of protein synthesis by catalyzing the GDP-GTP exchange on eIF2 to enable binding of methionyl-transfer-RNA to the ribosome. Despite the essential role of eIF2B in all cells, its defect curiously causes selective damage of white matter and in some cases damage to the ovaries alone. The ability of glia to regulate eIF2 activity may represent a critical protective mechanism in response to stress conditions.
Fabry disease
Fabry disease is a progressive X-linked lysosomal disorder due to a deficiency of the enzyme alpha-galactosidase A, leading to an accumulation of glycosphingolipids, mainly globotriaosylceramide GL-3 in lysosomes. This accumulation triggers tissue ischemia and fibrosis. The classic form of the disease presenting in males with no detectable enzyme activity, is characterized by angiokeratomas, acroparesthesia, hyperhidrosis, corneal opacity in childhood or adolescence and progressive vascular disease of the heart, kidneys, and central nervous system. MRI findings include white matter abnormalities and vertebrobasilar stroke. In contrast, patients with mild forms of Fabry disease (female carriers and males with residual alpha-galactosidase activity) may remain asymptomatic until late adulthood. The incidence of Fabry disease is estimated to be 1/100,000; however, with the advent of newborn screening the true incidence will be determined. Recently enzyme replacement therapy and pharmacological chaperone therapy have been introduced to lower the GL-3 accumulation in the lysosome.
Fucosidosis
Fucosidosis is a rare autosomal recessive disorder characterized by deficiency of the lysosomal enzyme alpha-L-fucosidase, which is required to break down (metabolize) certain complex compounds (e.g., fucose-containing glycolipids or fucose-containing glycoproteins). Fucose is a type of the sugar required by the body to perform certain functions (essential sugar). The inability to breakdown fucose-containing compounds results in their accumulation in various tissues in the body. Fucosidosis results in progressive neurological deterioration, skin abnormalities, delayed growth, skeletal disease and coarsening of facial features. The symptoms and severity of fucosidosis are highly variable and the disorder represents a disease spectrum in which individuals with mild cases have been known to live into the third or fourth decades. Individuals with severe cases of fucosidosis can develop life-threatening complications early in childhood. Hypomyelination is present on the MRI scans.
The disorder belongs to a group of diseases known as lysosomal storage disorders. Lysosomes are particles bound in membranes within cells that function as the primary digestive units within cells. Enzymes within lysosomes break down or digest particular nutrients, such as certain fats and carbohydrates. Low levels or inactivity of the alpha-L-fucosidase enzyme leads to the abnormal accumulation of fucose-containing compounds in the tissues of individuals with fucosidosis.
GM1 gangliosidosis
GM1 gangliosidosis is an autosomal recessive disorder due to deficiency of the lysosomal enzyme ß-galactosidase associated with mutations in the GLB1 gene. More than 100 mutations have been described. ß-galactosidase hydrolyses the ß-galactosyl residue from GM1 ganglioside, glycoproteins, and glycosaminoglycans. Deficiency of ß-galactosidase results in lysosomal storage of these substances, particularly in the central nervous system (CNS). Three types of GM1 gangliosidosis have been described. Type 1 or infantile GM1 gangliosidosis has its onset before 6 months of age with rapidly progressive hypotonia (low body tone) and CNS deterioration resulting in death by 1 to 2 years of age. Type II or late-infantile/ juvenile GM1 gangliosidosis presents with delay in cognitive and motor development between 7 months and 3 years of age with slow progression. Adult-onset GM1 gangliosidosis presents between 3 to 30 years of age with a progressive extrapyramidal disorder. MRI findings include delayed myelination, diffuse white matter abnormalities and abnormal signal in the basal ganglia.
L-2-hydroxyglutaric aciduria
L-2-hydroxyglutaric aciduria is a rare autosomal recessive disorder. Mutations in both copies of the L2HDGH gene result in deficiency of L-2-hydroxyglutarate dehydrogenase activity. L-2 hydroxyglutarate dehydrogenase is an FAD-linked mitochondrial enzyme that converts L-2 hydroxyglutarate to a-ketoglutarate. Biochemically, L-2-hydroxyglutaric aciduria presents with significantly elevated levels of L-2-hydroxyglutaric acid in the urine and CSF. Plasma amino acids reveal elevated lysine. Clinically, L-2 hydroxyglutaric aciduria presents with variable degrees of psychomotor and speech delay followed by a slowly progressive neurodegenerative disorder with cognitive decline. The MRI demonstrate a complex but characteristic pattern of abnormal signal intensity in the subcortical white matter bilaterally with frontal predominance and involvement of the globus pallidus, caudate and putamen bilaterally as well as the dentate nucleusAn increased risk of brain tumors has been described.
Megalencephalic leukoencephalopathy with subcortical cysts
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is an autosomal recessive condition which initially presents with macrocephaly (enlarged head size). Mild motor delay is followed by gradual motor deterioration with ataxia and spasticity. Cognitive abilities are relatively spared but seizures may occur in this classical form. Recessive MLC1 mutations are observed in 80% of patients with megalencephalic leukoencephalopathy with subcortical cysts (MLC). Other patients with the classical, deteriorating phenotype have two mutations in the HEPACAM gene. An improving phenotype has been described in patients with only one mutation in HEPACAM. Most parents with a single mutation had macrocephaly, indicating dominant inheritance. In some families with dominant HEPACAM mutations, the clinical picture and magnetic resonance imaging normalized, indicating that HEPACAM mutations can cause benign familial macrocephaly. In other families with dominant HEPACAM mutations, patients had macrocephaly and intellectual disability with or without autism. Diffuse white matter abnormalities on MRI are accompanied by anterior temporal cysts.
Multiple sulfatase deficiency
Multiple sulfatase deficiency (MSD) is a very rare leukodystrophy in which all of the known sulfatase enzymes (thought to be seven in number) are deficient or inoperative due to mutations in the SUMF1 gene. Major symptoms include mildly coarsened facial features, deafness, and an enlarged liver and spleen (hepatosplenomegaly). Abnormalities of the skeleton may occur, such as curvature of the spine (lumbar kyphosis) and the breast bone. The skin is usually dry and scaly (ichthyosis). Before symptoms are noticeable, children with this disorder usually develop more slowly than normal. They may not learn to walk or speak as quickly as other children.
Similar to metachromatic leukodystrophy, multiple sulfatase deficiency patients exhibit neurodegenerative disease in early childhood due to central nervous system (CNS) and peripheral demyelination with loss of sensory and motor functions. They also develop intellectual disability, hepatosplenomegaly, coarse facies, and corneal clouding as seen in patients with mucopolysaccharidoses. Ichthyosis and skeletal changes reflect enzyme deficiencies of steroid sulfatase (X-linked ichthyosis) and arylsulfatase E (chondrodysplasia punctata), respectively. The unique combination of neurodegeneration, coarse facial features, hepatosplenomegaly, and ichthyosis is not seen in other neuro-ichthyotic disorders. However, the sequential appearance of these clinical signs often delays the diagnosis of multiple sulfatase deficiency (MSD).
Pelizaeus-Merzbacher disease
Pelizaeus-Merzbacher disease (PMD), also known as X-linked spastic paraplegia, is a rare inherited disorder affecting the central nervous system that is associated with a lack of myelin sheath. Many areas of the central nervous system may be affected, including the deep portions of the cerebrum (subcortical), cerebellum, and/or brain stem. Symptoms may include the impaired ability to coordinate movement (ataxia), involuntary muscle spasms (spasticity) that result in slow, stiff movements of the legs, delays in reaching developmental milestones, loss of motor abilities, and the progressive deterioration of intellectual function. The symptoms of Pelizaeus-Merzbacher disease are usually slowly progressive. Several forms of the disorder have been identified, including classical X-linked Pelizaeus-Merzbacher disease; acute infantile (or connatal) Pelizaeus-Merzbacher disease; and adult-onset (or late-onset) Pelizaeus-Merzbacher disease. Various types of mutations of the X-linked proteolipid protein 1 gene (PLP1) that include copy number changes, point mutations, and insertions or deletions of a few bases lead to a clinical spectrum from the most severe connatal Pelizaeus-Merzbacher disease, to the least severe spastic paraplegia 2 (SPG2). The most common form of Pelizaeus-Merzbacher disease is caused by a duplication of the PLP1 gene and affects males. Signs of Pelizaeus-Merzbacher disease include nystagmus, hypotonia, tremors, titubation, ataxia, spasticity, athetotic movements and cognitive impairment; the major findings in SPG2 are leg weakness and spasticity. Supportive therapy for patients with PMD/SPG2 includes medications for seizures and spasticity; physical therapy, exercise, and orthotics for spasticity management; surgery for contractures and scoliosis; gastrostomy for severe dysphagia; proper wheelchair seating, physical therapy, and orthotics to prevent or ameliorate the effects of scoliosis; special education; and assistive communication devices.
An autosomal recessive condition clinically resembling classical Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher disease-like disease, has been described due to mutations in gap junction protein (GJA12). This condition affects both males and females.
Pol III-Related Leukodystrophies
The Pol III-related leukodystrophies comprise a group of 5 overlapping clinically defined hypomyelinating leukodystrophies including: Hypomyelination, hypodontia, hypogonadotropic hypogonadism (4H syndrome); Ataxia, delayed dentition, and hypomyelination (ADDH); Tremor-ataxia with central hypomyelination (TACH);Leukodystrophy with oligodontia (LO); and Hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum (HCAHC). These conditions present with varying combinations of motor dysfunction, abnormal teeth and hypogonadotropic hypogonadism. The MRI scan of the brain demonstrates hypomyelination. The condition is associated with autosomal recessive mutations in POLR3A or POLR3B.
Refsum disease
Refsum disease, also called hereditary sensory motor neuropathy type IV, is an autosomal recessive leukodystrophy in which the myelin sheath fails to grow. The disorder is caused by the accumulation of a methyl branched chain fatty acid (phytanic acid) in blood plasma and tissues due to mutations in the PHYH gene that encodes the peroxisomal enzyme phytanoyl-CoA hydroxylase that is responsible for the a-oxidation of phytanic acid. 90% of patients with Refsum disease have a mutation in the PHYH gene; whereas the remaining 10% have a mutation in the peroxisomal gene, Pex7, which is necessary for import of phytanoyl-CoA hydroxylase into peroxisomes. Refsum disease is characterized by progressive loss of vision (retinitis pigmentosa); degenerative nerve disease (peripheral neuropathy); failure of muscle coordination (ataxia); and dry, rough, scaly skin (ichthyosis). Treatment with a diet low in phytanic acid and avoidance of foods such as cold water fish, dairy and ruminant meats that contain phytanic acid can be beneficial. Plasmapheresis and the intestinal lipase inhibitor, Orlistat have shown some efficacy in lowering phytanic acid levels. However these therapies, while successful at diminishing the neurological symptoms do not prevent the slow progression of retinitis pigmentosa.
Salla disease
Salla disease is a rare autosomal recessive disorder due to deficiency of the sialic acid transporter, SLC17A5. Free sialic acid (N-acetylneuraminic acid) accumulates in lysosomes in various tissues. The severe form, infantile free sialic acid storage disorder, results in early death. Salla’s disease, which is more common in patients of Finnish descent, has wide clinical variability. Most children present between 3 and 9 months of age with hypotonia, ataxia, delayed motor milestones, and transient nystagmus. Cognitive delay and slow motor decline occurs after the second to third decade. Peripheral neuropathy may be present and contribute to motor disability. MRI findings are consistent with hypomyelination with minimal or extremely slow myelination. Myelin is present in the internal capsule and is usually normal in the cerebellum. The corpus callosum is usually thin. Treatment for Salla’s disease is supportive.
Sjögren-Larsson syndrome
Sjögren-Larsson syndrome (SLS) is caused by mutations in the ALDH3A2 gene that codes for fatty aldehyde dehydrogenase is located on chromosome 17p11.2. More than 70 different mutations in the ALDH3A2 gene have been identified in Sjögren-Larsson syndrome patients originating from about 120 different families. Fatty aldehyde dehydrogenase is necessary for the oxidation of long-chain aldehydes and alcohols to fatty acids. Deficiency of this enzyme leads to accumulation of these lipids leading to increased inflammatory lipids, the leukotrienes, in skin and brain, which are thought to be directly responsible for the symptoms of ichthyosis and delay in myelination. About 70% of Sjögren-Larsson syndrome patients are born preterm most likely due to the fetal excretion of abnormal lipids and leukotrienes causing inflammation and early labor. During early childhood (1–2 years of age) intellectual and motor disabilities gradually become clear, however, the typical MRI and H-MRS abnormalities, as well as crystalline maculopathy, may be absent, and normal radiologic and ocular findings do not exclude Sjögren-Larsson syndrome at this stage. Later on in childhood (from 3 years of age), the full-blown phenotype of Sjögren-Larsson syndrome with the classical triad of ichthyosis, spasticity, and intellectual disability is present with the typical findings of ophthalmological and MRI/H-MRS studies. Therapies consist of preventing skin lesions through application of special creams and urea-containing emollients and physical therapy and bracing to diminish contractures. Therapies to reduce the levels of leukotrienes, to prevent the skin lesions and improve neurological functioning are being studied.
X-linked adrenoleukodystrophy
X-linked adrenoleukodystrophy (ALD) is the most common leukodystrophy and affects the myelin or white matter of the brain and the spinal cord as well as the adrenal cortex. The gene for X-linked adrenoleukodystrophy, the ABCD1 gene, is located at Xq28 and encodes a peroxisomal protein belonging to the ATPase Binding Cassette proteins. There have been more than 1000 mutations reported in the ABCD1 gene (www.x-ald.nl). X-linked adrenoleukodystrophy is a progressive disease characterized by an accumulation of very long chain fatty acids, mainly of 26 carbons in chain length. There are several phenotypes of X-linked adrenoleukodystrophy, each distinguished by the age of onset and by the features that are present. All phenotypes can occur in the same kindred with 31-35% of affected males having the demyelinating childhood cerebral form (CCER) with typical onset between 4 and 8 yrs. Boys develop normally until the onset of cognitive decline and progressive neurologic deficits which lead to a vegetative state, blindness, seizures and death often within 3 yrs. Forty to 46% of males with X-linked adrenoleukodystrophy present in early adulthood with slowly progressive paraparesis (weakness and spasticity), sensory, and sphincter disturbances involving spinal cord long tracts. This form is called adrenomyeloneuropathy (AMN). At least 30% of men with adrenomyeloneuropathy (AMN) develop cerebral involvement that is similar to demyelinating childhood cerebral form (CCER). Fifty per cent of heterozygous females (carriers) develop overt neurologic disturbances resembling adrenomyeloneuropathy (AMN), with a mean age of onset of 37 yrs. The minimum frequency of hemizygotes (i.e., affected males) identified in the United States is estimated at 1:21,000 and that of hemizygotes plus heterozygotes (i.e., carrier females) 1:16,800.
Untreated adrenal insufficiency can be fatal and occurs independent of neurological symptoms. Earlier onset of demyelinating childhood cerebral form (CCER) correlates with more severe, rapidly progressive clinical manifestations. Boys with parieto-occipital lobe disease demonstrate visual and/or auditory processing abnormalities, impaired communication skills and gait disturbances prior to death. Boys with frontal lobe involvement have signs/symptoms similar to ADHD and are often misdiagnosed prior to death. The extent of demyelination can be quantitated using the MRI severity score of Loes.
X-linked adrenoleukodystrophy (ALD) in boys can be diagnosed by analysis of the very long chain fatty acids in plasma and if positive, mutation analysis of the ABCD1 gene is recommended. For females at risk of X-linked adrenoleukodystrophy, the most accurate test is targeted analysis of the family mutation in the ABCD1 gene as the plasma very long chain fatty acid test for females has a 20% false negative rate due to lyonization (selective X-inactivation) of the X-chromosome. It is important to screen all at-risk relatives for X-linked adrenoleukodystrophy as the males with X-linked adrenoleukodystrophy are at risk for Addison disease which is treatable with life-saving hormone therapy. Dietary therapy with Lorenzo’s oil if started early before MRI abnormalities occur and if plasma levels of very long chain fatty acids are normalized, has shown to statistically lower the development of demyelinating childhood cerebral form (CCER). Over one third of X-linked adrenoleukodystrophy boys will develop demyelinating childhood cerebral form (CCER) thus X-linked adrenoleukodystrophy boys who are diagnosed before neurological symptoms occur should be followed by a pediatric neurologist and have MRI every 6 months. At first signs of progressive white matter abnormalities on MRI, bone marrow transplantation, or hematopoetic cell transplantation, is recommended as the only effective long-term treatment for demyelinating childhood cerebral form (CCER); however, to achieve optimal survival and clinical outcomes, hematopoetic cell transplantation must occur prior to manifestations of symptoms. Gene therapy experimental treatment has been shown to be safe and efficacious.
With the development of a newborn screening test for X-linked adrenoleukodystrophy (ALD) all boys with X-linked adrenoleukodystrophy (ALD) will be diagnosed at an age before Addison disease and brain dysfunction occur. Thus life-saving therapies can be implemented early and other at risk relatives identified.
Zellweger syndrome spectrum disorders
Zellweger syndrome spectrum disorders, also known as peroxisomal biogenesis disorders (PBDs), are characterized by a deficiency or absence of peroxisomes in the cells of the liver, kidneys, and brain. Peroxisomes are very small, membrane-bound structures within the cytoplasm of cells that function as part of the body’s waste disposal system. In the absence of the enzymes normally found in peroxisomes, waste products, especially very long chain fatty acids, accumulate in the cells of the affected organ. The accumulation of these waste products has profound effects on the development of the fetus. Peroxisomal biogenesis disorders are inherited as autosomal recessive disorders and have two clinically distinct subtypes: the Zellweger syndrome spectrum (ZSS) disorders and rhizomelic chondrodysplasia punctata (RCDP) type 1. Peroxisomal biogenesis disorders are caused by defects in any of at least 14 different PEX genes, which encode proteins involved in peroxisome assembly and proliferation. There is genetic heterogeneity among peroxisomal biogenesis disorders and this is present in all defective PEX genes. The peroxisomal biogenesis disorders with the mildest phenotype are known by the clinical names, neonatal adrenoleukodystrophy and infantile Refsum’s disease. A range of symptoms are seen including developmental delay, sensorineural hearing loss, visual abnormalities, adrenal insufficiency and liver dysfunction. MRI scans may show developmental abnormalities of the brain and progressive white matter changes may develop. Diagnosis of peroxisomal biogenesis disorders is made by finding an increase in the plasma very long chain fatty acids and the branched chain fatty acids, phytanic and pristanic. Additional biochemical laboratory tests are the measurement of red blood cell plasmalogens.
Krabbe leukodystrophy
Krabbe disease also called globoid cell leukodystrophy is a severe neurological condition. It is part of a group of disorders known as leukodystrophies, which result from the loss of myelin (demyelination) in the nervous system. Krabbe disease (globoid cell leukodystrophy) is also characterized by abnormal cells in the brain called globoid cells, which are large cells that usually have more than one nucleus.
In the United States, Krabbe disease (globoid cell leukodystrophy) affects about 1 in 100,000 individuals. A higher incidence (6 cases per 1,000 people) has been reported in a few isolated communities in Israel 11).
The most common form of Krabbe disease (globoid cell leukodystrophy), called the infantile form, usually begins before the age of 1. Initial signs and symptoms typically include irritability, muscle weakness, feeding difficulties, episodes of fever without any sign of infection, stiff posture, and delayed mental and physical development. As the disease progresses, muscles continue to weaken, affecting the infant’s ability to move, chew, swallow, and breathe. Affected infants also experience vision loss and seizures. Because of the severity of the condition, individuals with the infantile form of Krabbe disease rarely survive beyond the age of 2.
Less commonly, Krabbe disease begins in childhood, adolescence, or adulthood (late-onset forms). Vision problems and walking difficulties are the most common initial symptoms in these forms of the disorder, however, signs and symptoms vary considerably among affected individuals. Individuals with late-onset Krabbe disease may survive many years after the condition begins.
Krabbe leukodystrophy causes
Mutations in the galactocerebrosidase (GALC) gene cause Krabbe disease. This gene provides instructions for making an enzyme called galactosylceramidase, which breaks down certain fats called galactolipids. One galactolipid broken down by galactosylceramidase, called galactosylceramide, is an important component of myelin. Breakdown of galactosylceramide is part of the normal turnover of myelin that occurs throughout life. Another galactolipid, called psychosine, which is formed during the production of myelin, is toxic if not broken down by galactosylceramidase.
GALC gene mutations severely reduce the activity of the galactosylceramidase enzyme. As a result, galactosylceramide and psychosine cannot be broken down. Excess galactosylceramide accumulates in certain cells, forming globoid cells. The accumulation of these galactolipids causes damage to myelin-forming cells, which impairs the formation of myelin and leads to demyelination in the nervous system. Without myelin, nerves in the brain and other parts of the body cannot transmit signals properly, leading to the signs and symptoms of Krabbe disease.
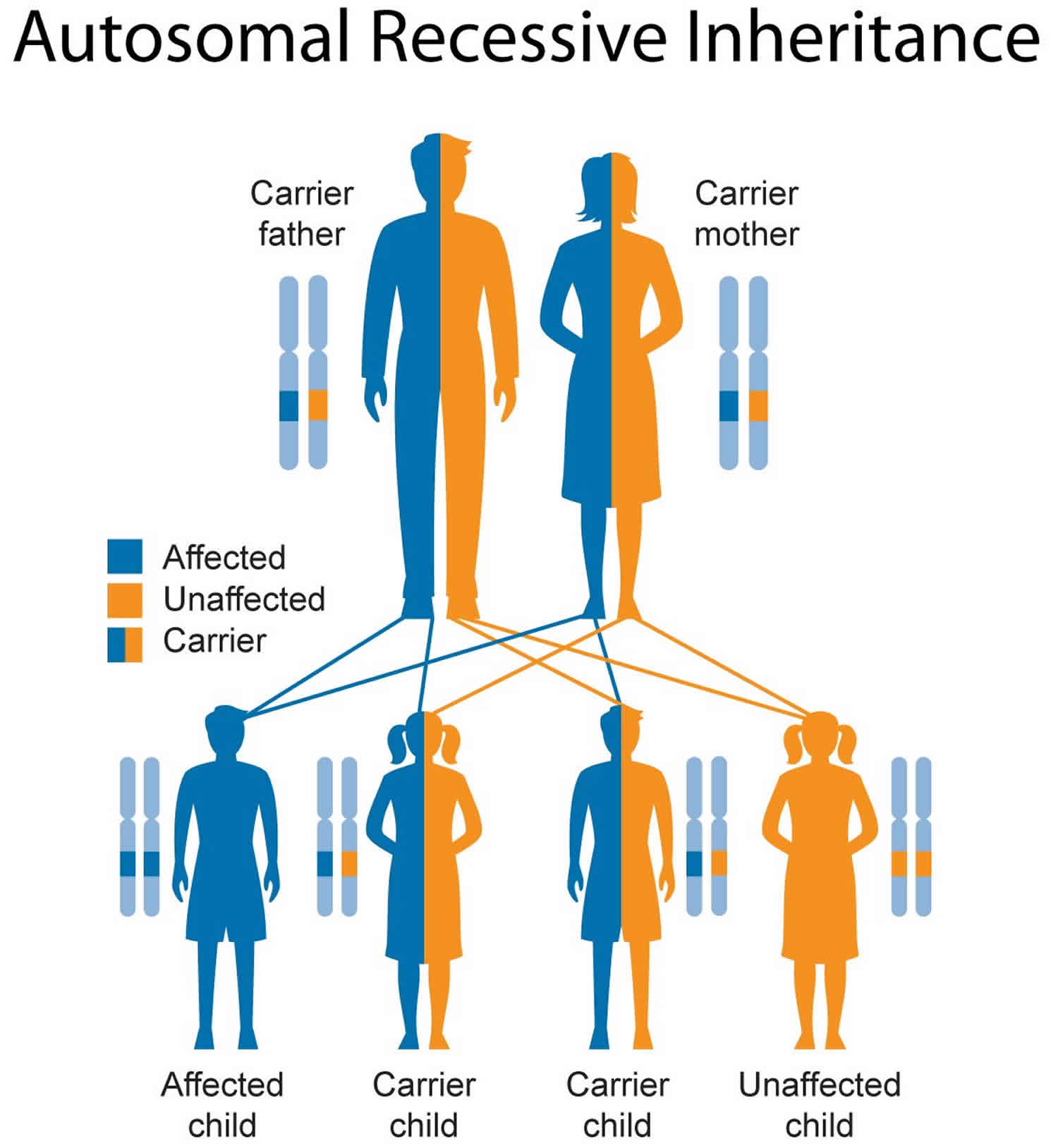
Krabbe leukodystrophy inheritance pattern
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Genetic counseling may help you understand the risks of passing leukodystrophy on to any children you have.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Figure 4. Krabbe leukodystrophy autosomal recessive inheritance pattern
Krabbe leukodystrophy symptoms
The majority of cases of Krabbe disease appear within the first year of life. The patients rapidly regress to a condition with little to no brain function, and generally die by age 2, though some have lived longer. Death generally occurs as a result of a respiratory infection or brain fever. Symptoms that might be encountered in the infantile form of Krabbe disease include:
- Developmental delay
- Seizures
- Limb stiffness
- Optic atrophy: wasting of a muscle of the eye, resulting in vision diffculties
- Neurosensoral deafness
- Extreme irritability
- Spasticity: presence of spasms
- Ataxia: loss of the ability to control muscular movement
- Progressive psychomotor decline: progressive decline in the coordination of movement
Although the majority of Krabbe Disease patients show symptoms within the first year of life, there have been cases diagnosed at all ages, through late adulthood. In general, the earlier the diagnosis, the more rapid the progression of the disease. Those who first show symptoms at ages 2-14 will regress and become severely incapacitated, and generally die 2-7 years following diagnosis. Some patients who have been diagnosed in the adolescent and adult years have symptoms that remain confined to weakness without any intellectual deterioration, while others may become bedridden and deteriorate both mentally and physically.
Krabbe leukodystrophy diagnosis
Krabbe Disease can be diagnosed by a biochemical assay that measures the galactocerebrosidase (GALC) activity from a blood sample or skin biopsy. It should be noted that the absolute level of GALC activity is not an indicator of prognosis; that is, a particularly low GALC activity does not necessarily predict a more rapid progression of disease than a somewhat higher GALC activity.
The genetic basis for Krabbe disease is known, so it also may be possible to perform DNA sequencing of the gene in order to confirm the diagnosis of Krabbe disease. In conjunction with genetic counseling, this knowledge may also allow relatives of patients with Krabbe disease to be tested for the presence of the genetic mutation responsible, allowing them to make informed decisions about having children.
Krabbe leukodystrophy treatment
Most treatment of Krabbe Disease is supportive. However, one medical treatment that has been demonstrated to have some effect is hematopoietic stem cell transplant. This method appears to be of some benefit in cases of later onset or in infantile patients who have been diagnosed before or at birth. The clinical course of patients who have received the transplants seems to be less severe, and an improvement in the pathology of the disease can be seen by MRI. However, HSCT does not appear to be beneficial in the case of infantile patients who have already begun displaying the symptoms of Krabbe Disease. Hematopoietic stem cell transplant has been attempted on three fetuses with Krabbe Disease, and failed in all cases, presumably because the donor cells were not sufficiently engrafted.
One promising treatment is genetic therapy, where the deficient gene (GALC) is delivered in a harmless virus. Another promising method of treatment is stem cell therapy, which can provide healthy cells with GALC activity to allow for remyelination. However, none of these methods have yet been attempted on human subjects.
Metachromatic leukodystrophy
Metachromatic leukodystrophy is an inherited disorder characterized by the accumulation of fats called sulfatides in cells 12). Metachromatic leukodystrophy gets its name from the way cells with an accumulation of sulfatides appear when viewed under a microscope. The sulfatides form granules that are described as metachromatic, which means they pick up color differently than surrounding cellular material when stained for examination. This accumulation especially affects cells in the nervous system that produce myelin, the substance that insulates and protects nerves. Nerve cells covered by myelin make up a tissue called white matter. Sulfatide accumulation in myelin-producing cells causes progressive destruction of white matter (leukodystrophy) throughout the nervous system, including in the brain and spinal cord (the central nervous system) and the nerves connecting the brain and spinal cord to muscles and sensory cells that detect sensations such as touch, pain, heat, and sound (the peripheral nervous system).
In people with metachromatic leukodystrophy, white matter damage causes progressive deterioration of intellectual functions and motor skills, such as the ability to walk 13). Affected individuals also develop loss of sensation in the extremities (peripheral neuropathy), incontinence, seizures, paralysis, an inability to speak, blindness, and hearing loss. Eventually they lose awareness of their surroundings and become unresponsive. While neurological problems are the primary feature of metachromatic leukodystrophy, effects of sulfatide accumulation on other organs and tissues have been reported, most often involving the gallbladder.
Metachromatic leukodystrophy is reported to occur in 1 in 40,000 to 160,000 individuals worldwide 14). Metachromatic leukodystrophy is more common in certain genetically isolated populations: 1 in 75 in a small group of Jews who immigrated to Israel from southern Arabia (Habbanites), 1 in 2,500 in the western portion of the Navajo Nation, and 1 in 8,000 among Arab groups in Israel.
There are three forms of metachromatic leukodystrophy, defined by the age of onset of the disease. The late infantile form of metachromatic leukodystrophy is the most common, and produces symptoms between the ages of 1 and 2. The juvenile form generally becomes apparent between the ages of 4 and 12, and the adult form occurs after age 14. As with all the leukodystrophies, the symptoms can vary widely, although in all cases there is a progressive loss of physical and intellectual function over a relatively extended period of time. In general, the earlier the onset, the more rapid the progression of the disease.
Late infantile metachromatic leukodystrophy
The most common form of metachromatic leukodystrophy, affecting about 50 to 60 percent of all individuals with this disorder, is called the late infantile form 15). Late infantile form of metachromatic leukodystrophy usually appears in the second year of life. After a period of apparently normal growth and development, skills such as walking and speech may begin to deteriorate. Once clinical symptoms become noticeable, they often appear to progress rapidly over a period of several months, with alternating periods of stabilization and decline. The child eventually becomes bedridden, unable to speak or feed independently. There may be seizures at this stage, which eventually disappear. Contractures are common and apparently painful. The child is still able to smile and respond to parents at this stage, but eventually may become blind and largely unresponsive. Swallowing eventually becomes difficult and a feeding tube becomes necessary. Individuals with the late infantile form of metachromatic leukodystrophy typically do not survive past childhood 16). With modern treatment and care, the child may survive for 5-10 years.
Death generally occurs as the result of an infection such as pneumonia, as opposed to being a direct result of the metachromatic leukodystrophy. Other symptoms that may be encountered are listed below.
- Developmental delay
- Hypotonia: decreased muscle tone
- Esotropia: cross-eyed
- Psychomotor regression
- Clumsiness
- Spasticity: increased reflexes
- Nystagmus: type of abnormal eye movement
- Weakness
- Decreased speech
- Seizures
- Ataxia: loss of the ability to coordinate muscular movement
- Quadriplegia: paralysis from the neck down
- Eventual absence of voluntary functions
Juvenile metachromatic leukodystrophy
In 20 to 30 percent of individuals with metachromatic leukodystrophy, onset occurs between the age of 4 and adolescence. In this juvenile form, the first signs of the disorder may be behavioral problems and increasing difficulty with schoolwork. Progression of the juvenile form is slower than in the late infantile form, and affected individuals may survive for about 20 years after diagnosis 17). Symptoms may include incontinence, difficulties in walking and slurred speech. As symptoms advance, children may develop seizures, abnormal postures, tremor, and eventually lose the ability to walk. The final stages of the disease are similar to the late infantile form. An increasing number of patients are living into adulthood.
Adult metachromatic leukodystrophy
The adult form of metachromatic leukodystrophy affects approximately 15 to 20 percent of individuals with metachromatic leukodystrophy 18). In this form, the first symptoms appear during the teenage years (14 years) or later as late as 50 years 19). Often behavioral problems such as alcoholism, drug abuse, or difficulties at school or work are the first symptoms to appear. The affected individual may experience psychiatric symptoms such as delusions or hallucinations. People with the adult form of metachromatic leukodystrophy may survive for 20 to 30 years after diagnosis. During this time there may be some periods of relative stability and other periods of more rapid decline.
Metachromatic leukodystrophy causes
Most individuals with metachromatic leukodystrophy have mutations in the arylsulfatase A (ARSA) gene, which provides instructions for making the enzyme arylsulfatase A (also called sulfatide sulfatase). This enzyme is located in cellular structures called lysosomes, which are the cell’s recycling centers. Within lysosomes, arylsulfatase A helps break down sulfatides, also called glycolipid- cerebroside sulfates, which are fats present in myelin. When arylsulfatase A (ARSA) is deficient, the sulfatides build up in the myelin to high levels, disrupting the myelin structure and causing demyelination to occur in both the central nervous system and in the peripheral nervous system. The sulfatides will also build up in the visceral organs (such as the kidneys), and will be excreted at high levels in the urine.
A few individuals with metachromatic leukodystrophy have mutations in the prosaposin (PSAP) gene. This gene provides instructions for making a protein called prosaposin that is broken up (cleaved) into smaller proteins (saposin A, B, C, and D) that assist enzymes in breaking down various fats. One of these smaller proteins is called saposin B; this protein works with arylsulfatase A arylsulfatase A (ARSA) to break down sulfatides.
Mutations in the arylsulfatase A (ARSA) or prosaposin (PSAP) genes result in a decreased ability to break down sulfatides, resulting in the accumulation of these substances in cells. Excess sulfatides are toxic to the nervous system. The accumulation gradually destroys myelin-producing cells, leading to the impairment of nervous system function that occurs in metachromatic leukodystrophy.
In some cases, individuals with very low arylsulfatase A (ARSA) activity show no symptoms of metachromatic leukodystrophy. This condition is called pseudoarylsulfatase deficiency.
Metachromatic leukodystrophy inheritance pattern
Metachromatic leukodystrophy is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Genetic counseling may help you understand the risks of passing leukodystrophy on to any children you have.
- To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. Online directories are provided by the American College of Medical Genetics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) and the National Society of Genetic Counselors (https://www.findageneticcounselor.com/).
Figure 5. Metachromatic leukodystrophy autosomal recessive inheritance pattern
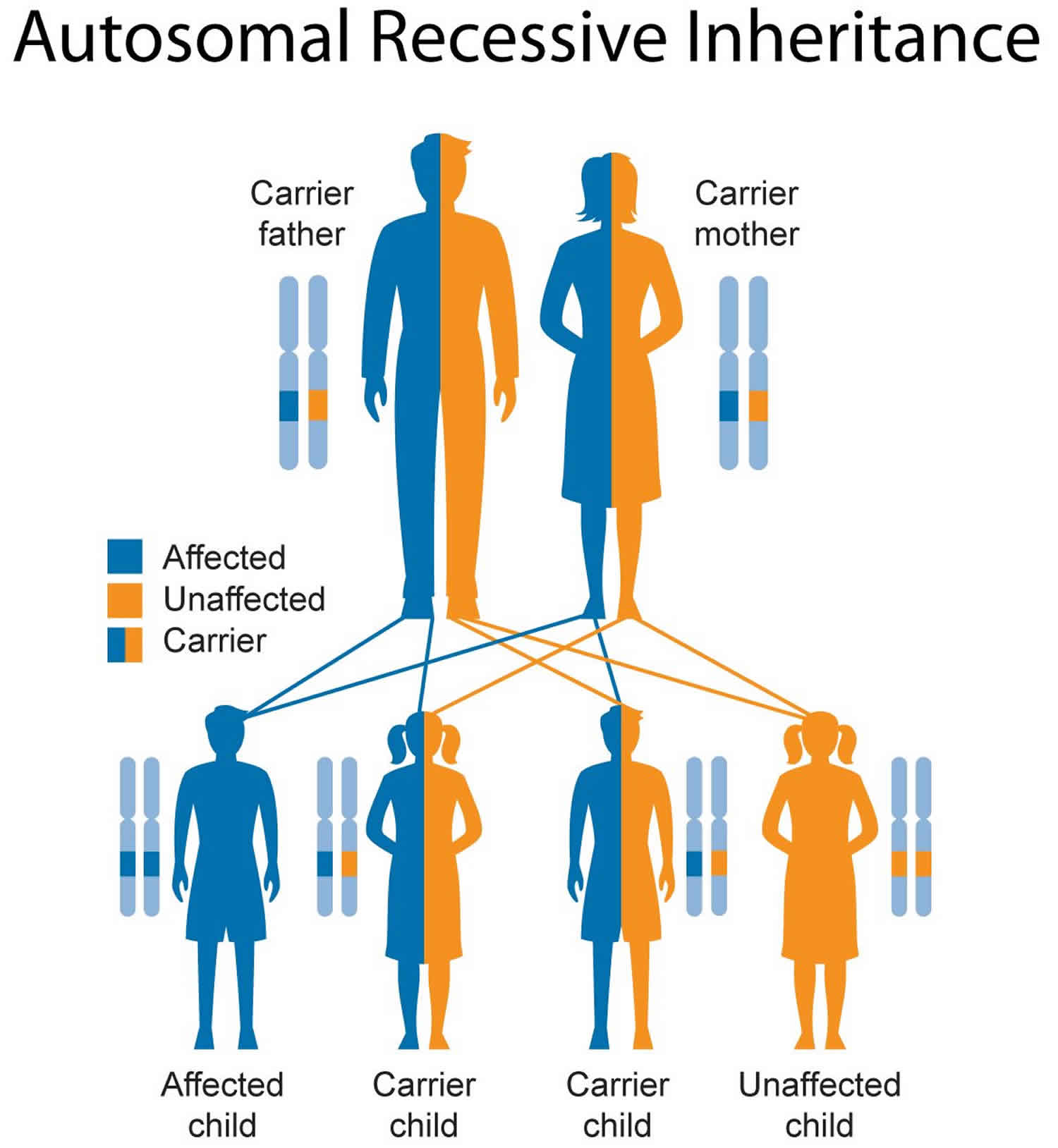
Metachromatic leukodystrophy diagnosis
If a child displays some of the symptoms described previously, a series of biochemical evaluations and brain imaging studies can be performed.
Biochemical evaluations
Because the most common cause of metachromatic leukodystrophy is a deficiency of arylsulfatase A (ARSA), a blood sample or skin punch biopsy may be taken, and arylsulfatase A (ARSA) activity can be measured; a low activity is suggestive of metachromatic leukodystrophy. However, it should be noted that low arylsulfatase A (ARSA) activity does not necessarily indicate metachromatic leukodystrophy. There is a mutation in the arylsulfatase A (ARSA) gene known as the “pseudodeficiency allele” that results in a lowering of arylsulfatase A (ARSA) activity. However, this pseudodeficiency allele does not directly cause metachromatic leukodystrophy. Roughly 10% of the population carries this pseudodeficiency allele, so biochemical results should be interpreted in conjunction with other tests. Other studies that may be performed include measurement of sulfatides in urine, a test for elevated cerebrospinal fluid protein, slowed nerve conduction, and changes in electrical potential that may be indicative of leukodystrophy.
Prenatal diagnosis for metachromatic leukodystrophy is available.
Brain imaging studies
An MRI (Magnetic Resonance Imaging) may be performed to look for white matter disturbances characteristic of metachromatic leukodystrophy.
Metachromatic leukodystrophy treatment
Currently, the only treatment for metachromatic leukodystrophy is bone marrow transplantation; this means that cells that produce normal arylsulfatase A (ARSA) are introduced into the patient, and the normal arylsulfatase A (ARSA) protein is then taken up into the deficient cells, allowing sulfatides in those cells to be broken down. However, this is only useful for those who are pre-symptomatic or those with very mild neurological manifestations. This highlights the importance of testing asymptomatic brothers and sisters of patients who have metachromatic leukodystrophy. This treatment can slow the disease progress and increase the quality of life for the patient 20).
Leukodystrophy prognosis
The prognosis for the leukodystrophies varies according to the specific type of leukodystrophy.
Leukodystrophy life expectancy
Life expectancy for leukodystrophies varies according to the specific type of leukodystrophy.
Leukodystrophy symptoms
Involvement of the white matter tracts almost universally leads to motor involvement that manifests as hypotonia in early childhood and progresses to spasticity over time. This may lead to variable motor impairment, from mild spastic diplegia to severe spastic quadriplegia that limits purposeful movement. In addition, motor dysfunction is likely to significantly impair vital functions including swallowing, chewing and (in some cases) respiration. Spasticity may result in orthopedic complications such as scoliosis and large joint luxation.
Significant pyramidal dysfunction (i.e., spasticity) may sometimes mask or overshadow the presence of extrapyramidal movement disorders such as dystonia and/or dyskinesias. For example, in MCT8-specific thyroid hormone cell transporter deficiency dystonia is a prominent finding.
Ataxia is a predominant finding in some leukodystrophies and can be disabling; for example, childhood ataxia with central nervous system hypomyelination/vanishing white matter and hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome).
Seizures are an often late manifestation of leukodystrophies, with the exception of rare leukodystrophies (e.g., Alexander disease) in which they are often a presenting feature.
Delay in cognitive development or change in cognitive function over time, while far less pronounced than motor dysfunction, can be common in the child or adult with leukodystrophy. Because progressive loss of cognitive function is slow in the majority of leukodystrophies, dementia is not an early feature.
Leukodystrophy treatment
Although the underlying mechanisms of leukodystrophies are diverse, many symptoms are similar across this group of disorders. In the great majority of cases, primary treatment is not possible, but management of symptoms can improve the comfort and care of individuals with these complex disorders.
Ideally, the child or adult with a leukodystrophy is managed in a multidisciplinary setting by providers experienced in the care of persons with a leukodystrophy.
Spasticity
- Pharmacologic agents are used to manage muscle tone and block neuronal signaling to muscle (chemodenervaton). Intensive physical therapy is used to improve mobility and function.
Extrapyramidal manifestations
- Dystonia and dyskinesias may cause significant disability; pharmacologic treatment may result in significant functional improvement.
Ataxia
- No specific treatment of ataxia exists, although rehabilitative measures can be of great assistance.
Seizures
- Seizures should be treated with typical anticonvulsants and are rarely refractory, except on occasion at the end of life.
Cognitive developmental delay/encephalopathy
- It is important to advocate for persons with a leukodystrophy in school or at work to avoid limitations related to their motor disabilities. Augmentative communication may be used to address speech deficits. Accommodations for cognitive delays and fine motor disabilities should be used as needed.
Orthopedic
- Attention should be given to the prevention and treatment of orthopedic problems, such as hip dislocation and scoliosis.
Feeding
Swallowing dysfunction and pulmonary problems resulting from the increased risk of aspiration are common as the disease progresses. Decreased nutritional intake and failure to thrive may also occur. The decision to place a gastrostomy tube for nutrition is based on the overall health status of the individual, expected disease course, and family and patient wishes.
Prevention of Primary Manifestations
Primary disease manifestations can be prevented in a few of the leukodystrophies: in X-linked adrenoleukodystrophy, Krabbe disease, and metachromatic leukodystrophy, for example, hematopoietic stem cell transplantation or bone marrow transplantation may be beneficial if performed early in the disease course. Patients with these disorders should be referred to specialized centers for consideration of hematopoietic stem cell transplantation or bone marrow transplantation 21).
Surveillance
Standard surveillance includes the following:
- Routine measurement of weight and height to assess growth and nutritional status
- Physical examination and/or serial x-rays of the hips and spine to monitor for orthopedic complications
- Routine history on signs and symptoms of seizures
Certain disorders require specialized surveillance, for example monitoring for the development of hydrocephalus in Alexander disease.
Agents/Circumstances to Avoid
In a number of leukodystrophies anecdotal evidence suggests episodic worsening of manifestations with mild head injuries and infection. While this has been clearly documented only for childhood ataxia with central nervous system hypomyelination/vanishing white matter, it appears prudent to avoid these triggers when possible.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Genetic counseling may help you understand the risks of passing leukodystrophy on to any children you have.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
References [ + ]




{kind=link}