Contents
Juvenile polyposis syndrome
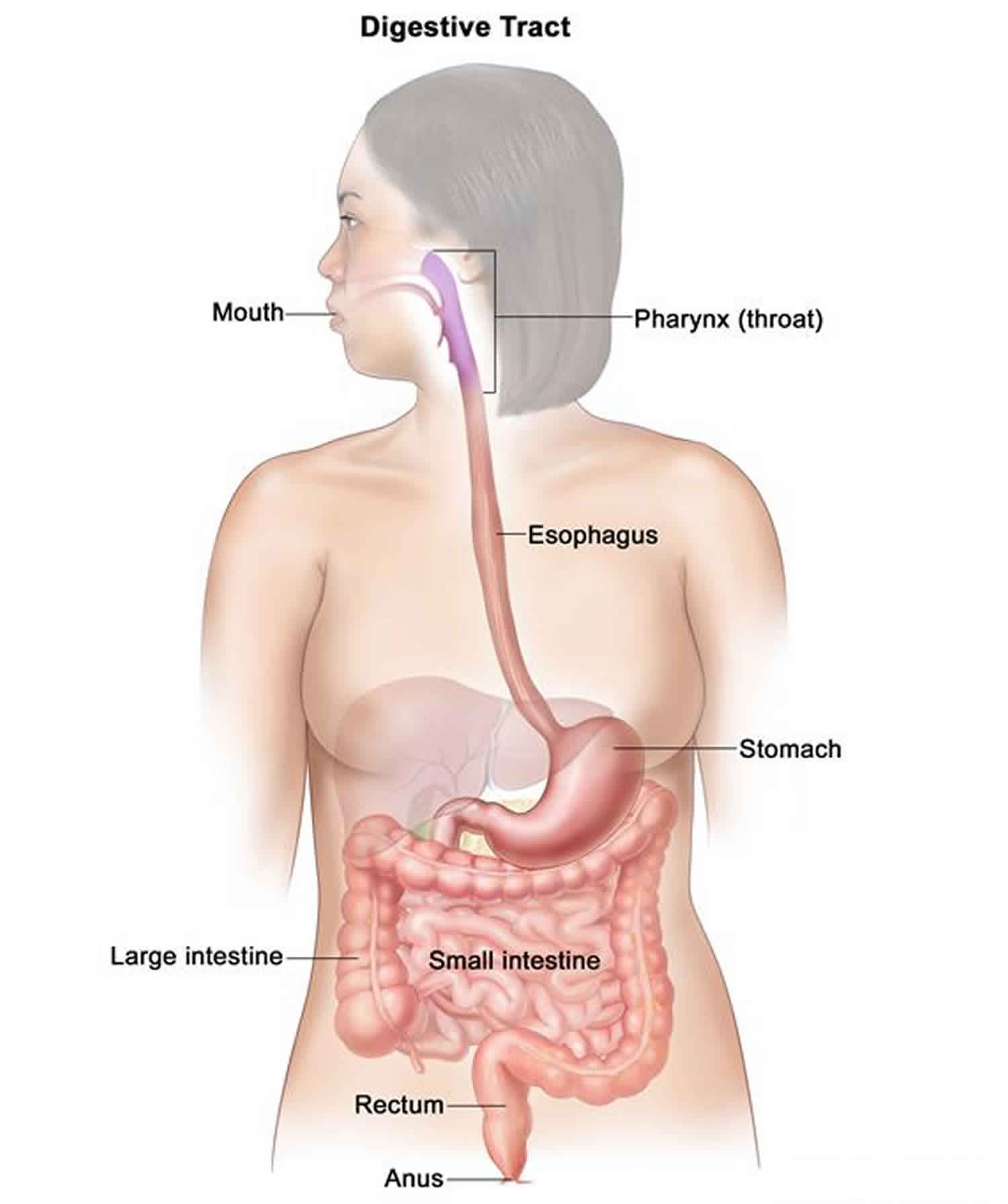
Juvenile polyposis syndrome also called juvenile intestinal polyposis, is a disorder characterized by multiple noncancerous (benign) growths called juvenile polyps. Most people with juvenile polyposis syndrome have some polyps by the age of age 20. The term “juvenile” is usually a misnomer as it is not related to the patient age of onset of polyp but the histopathology of the polyp itself. These growths occur in the gastrointestinal tract, typically in the large intestine (colon). The number of polyps varies from only a few to hundreds, even among affected members of the same family. Most juvenile polyps are benign and symptomatic ones usually present with gastrointestinal bleeding, a shortage of red blood cells (anemia), abdominal pain, diarrhea and/or bowel obstruction depending on the polyp burden in the gastrointestinal tract. Approximately 15 percent of people with juvenile polyposis syndrome have other abnormalities, such as a twisting of the intestines (intestinal malrotation), heart or brain abnormalities, an opening in the roof of the mouth (cleft palate), extra fingers or toes (polydactyly), and abnormalities of the genitalia or urinary tract.
Juvenile polyposis syndrome occurs in approximately 1 in 100,000 to 160,000 individuals worldwide 1).
Juvenile polyposis syndrome is most frequently caused by mutations in the SMAD4 or BMPR1A genes, but together these genes accounts for only 40% of cases 2). In 60 percent of patients, the underlying cause of juvenile polyposis syndrome is unknown.
Juvenile polyposis syndrome is diagnosed when a person has any one of the following: (1) more than five juvenile polyps of the colon or rectum; (2) juvenile polyps in other parts of the gastrointestinal tract; or (3) any number of juvenile polyps and one or more affected family members. Single juvenile polyps are relatively common in children and are not characteristic of juvenile polyposis syndrome.
Three types of juvenile polyposis syndrome have been described, based on the signs and symptoms of the disorder 3). Juvenile Polyposis of Infancy is characterized by polyps that occur throughout the gastrointestinal tract during infancy 4). Juvenile polyposis of infancy is the most severe form of the disorder and is associated with the poorest outcome. Children with this type may develop a condition called protein-losing enteropathy. Juvenile polyposis of infancy results in severe diarrhea, failure to gain weight and grow at the expected rate (failure to thrive), and general wasting and weight loss (cachexia). Another type called Generalized Juvenile Polyposis is diagnosed when polyps develop throughout the gastrointestinal tract 5). In the third type, known as Juvenile Polyposis Coli (colon involved only), affected individuals develop polyps only in their colon. People with generalized juvenile polyposis and juvenile polyposis coli typically develop polyps during childhood 6).
While the polyps associated with juvenile polyposis syndrome are most often benign, they can change into a malignant cancer. It is estimated that people with juvenile polyposis syndrome have a 9 to 50 percent risk of developing a cancer of the gastrointestinal tract. The most common type of cancer seen in people with juvenile polyposis syndrome is colorectal cancer, but cancers in other parts of the digestive system have also been described, such as cancers of the stomach, upper gastrointestinal tract and pancreas. The incidence of colorectal cancer in people with juvenile polyposis syndrome is 17%-22% by the age of 35 and as high as 68% by the age of 60. The incidence of gastric cancer in those with gastric polyps is 21 percent 7).
Management of juvenile polyposis syndrome includes routine colonoscopy with removal of any polyps to reduce the risk of bleeding, intestinal obstruction, and colon cancer. When the number of polyps is large, removal of all or part of the colon or stomach may become needed. Additional screening can include upper endoscopy, complete blood count, and monitoring for symptoms such as rectal bleeding and/or anemia abdominal pain, constipation, diarrhea, or change in stool size, shape, and/or color 8).
Combined juvenile polyposis syndrome and hereditary hemorrhagic telangiectasia syndrome
There is a condition related to juvenile polyposis syndrome that is also caused by alterations in the SMAD4 gene. It is not associated with alterations in the BMPR1A gene. This condition is known as combined juvenile polyposis and hereditary hemorrhagic telangiectasia syndrome (Osler-Weber-Rendu syndrome). It is estimated that 15-22 percent of people with a genetic alteration in SMAD4 may have combined juvenile polyposis syndrome and hereditary hemorrhagic telangiectasia syndrome.
In addition to the features of juvenile polyposis syndrome (gastrointestinal bleeding, gastric and colorectal polyps), individuals with combined juvenile polyposis syndrome and hereditary hemorrhagic telangiectasia syndrome can have variable features of another condition known as hereditary hemorrhagic telangiectasia, which include:
- Telangiectasias (small dilated blood vessels under the skin or mucous membranes)
- Arterio-venous malformations (AVMs) (larger blood vessel abnormalities) involving the lungs, liver, brain or gastrointestinal tract
- Epistaxis (nosebleeds)
- Intracranial bleeding
The features of hereditary hemorrhagic telangiectasia may manifest in early childhood. The frequency of each of these hereditary hemorrhagic telangiectasia-associated features in individuals with combined juvenile polyposis syndrome/hereditary hemorrhagic telangiectasia syndrome has not been well-established; however pulmonary arterio-venous malformations appear to be more common while nosebleeds and telangiectases have not been consistently reported in individuals with combined juvenile polyposis syndrome/hereditary hemorrhagic telangiectasia syndrome.
Juvenile polyposis syndrome causes
In 60 percent of patients, the underlying cause of juvenile polyposis syndrome is unknown. In the other 40 percent of patients, juvenile polyposis syndrome develops as the result of mutations in one of two different genes, BMPR1A and SMAD4. BMPR1A is located on chromosome 10 at position q22.3-23.2 and SMAD4 is located on chromosome 18 at position q21.1. These genes provide instructions for making proteins that are involved in transmitting chemical signals from the cell membrane to the nucleus. This type of signaling pathway allows the environment outside the cell to affect how the cell produces other proteins. The BMPR1A and SMAD4 proteins work together to help regulate the activity of particular genes and the growth and division (proliferation) of cells.
The BMPR1A gene produces a protein known as a cell surface receptor. This protein and the protein encoded by the SMAD4 gene act as tumor suppressors, which means that they help to keep cells from growing and dividing too quickly. They also promote cell death.
Mutations in the BMPR1A gene or the SMAD4 gene disrupt cell signaling and interfere with their roles in regulating gene activity and cell proliferation. This lack of regulation causes cells to grow and divide in an uncontrolled way, which can lead to polyp formation.
In children with juvenile polyposis syndrome, however, each cell contains only one working copy of either BMPR1A or SMAD4. While the second copy is present, it is mutated and does not function properly.
Children with only one working copy of either BMPR1A or SMAD4 are born and develop normally, but are at an increased risk to develop both non-cancerous and cancerous growths. These growths are believed to develop because, over time, the one remaining working copy of either the BMPR1A or SMAD4 genes may become altered within one or more cells. This can lead to abnormal growth of the affected cells, increasing their chance to form tumors or cancers.
Juvenile polyposis syndrome inheritance pattern
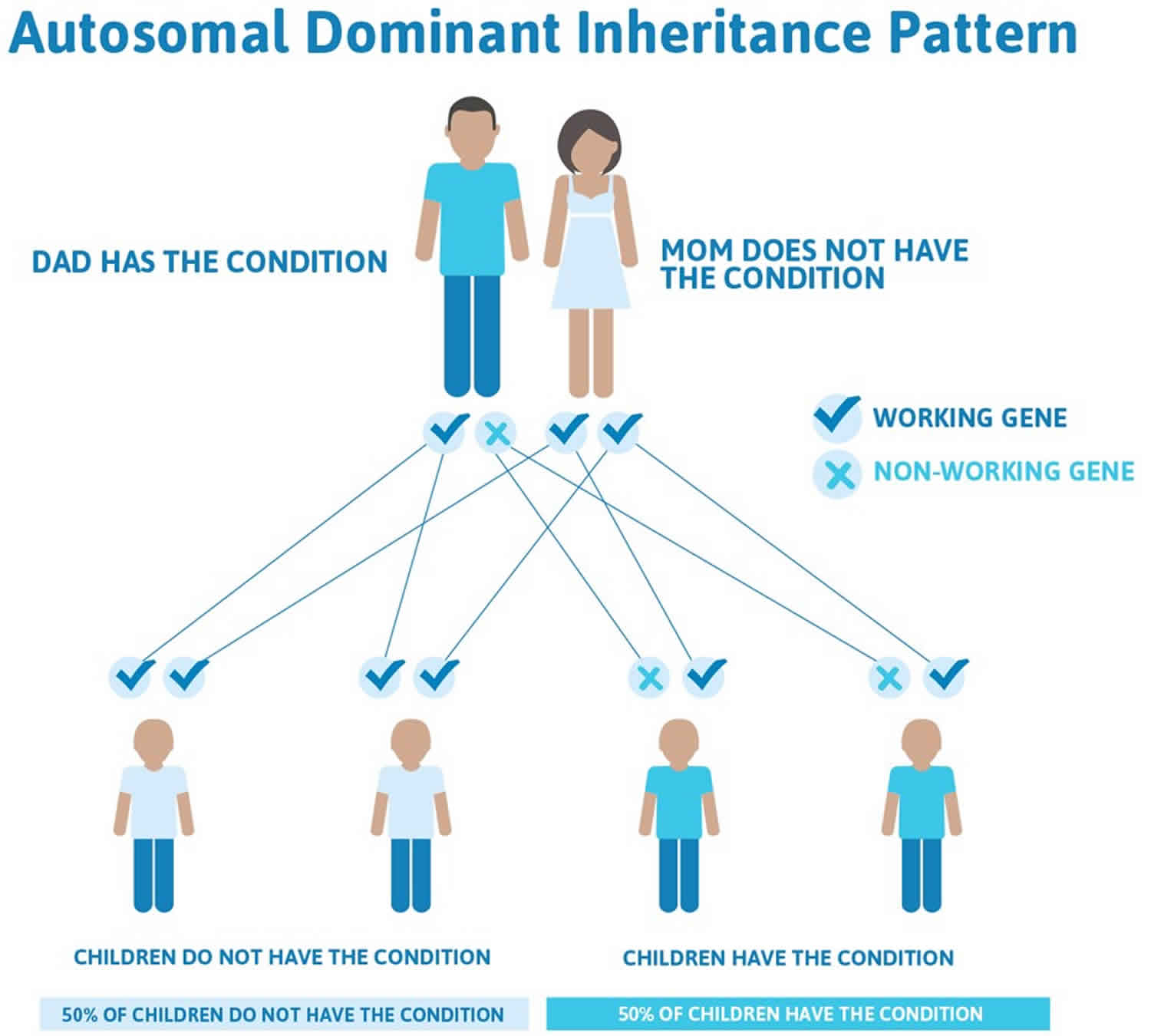
Juvenile polyposis syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. A person carrying an alteration in one copy of the BMPR1A gene or in one copy of the SMAD4 gene has a 50 percent chance of passing this same alteration on to each of his or her children. Children who inherit the altered gene copy will have juvenile polyposis syndrome and be at increased risk to develop polyps and other features associated with this condition.
In approximately 75 percent of patients with juvenile polyposis syndrome, an affected person inherits an altered copy of the BMPR1A or SMAD4 gene from a parent who also has juvenile polyposis syndrome. The remaining 25 percent of cases result from new mutations in the gene and occur in people with no history of the disorder in their family. In these individuals, juvenile polyposis syndrome likely results from the development of a new mutation in one copy of either BMPR1A or SMAD4. Although these individuals will be the first ones in their family to carry the genetic change, each of their future offspring will have a 50 percent chance of inheriting the genetic mutation.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 1. Juvenile polyposis syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Juvenile polyposis syndrome symptoms
Children may begin to experience symptoms during early childhood. Abnormal signs and symptoms that may develop in the course of juvenile polyposis syndrome include:
- rectal bleeding
- diarrhea
- anemia
- bowel obstruction
- visible rectal polyp
Most polyps in juvenile polyposis syndrome are non-cancerous (benign). However, polyps can change and become cancerous. Colon cancer is the most serious risk of juvenile polyposis syndrome, with up to a 50 percent chance of developing colon cancer during their lifetime.
Clinically, juvenile polyposis can present in two forms. The first is called juvenile polyposis of infancy. This is a generalized form occurring in infants with polyps in the stomach, small bowel and colon 9). The polyps vary in size from 1 to 30 mm and may be sessile or pedunculated. These infants suffer from diarrhea, hemorrhage, malnutrition and intussusception. Death usually occurs at an early age. In addition, many of these patients have congenital abnormalities, including enlarged head (macrocephaly) and generalized weak muscle tone (hypotonia) 10). Some investigators suggest that this rare form of juvenile polyposis is caused by continuous deletion of BMPR1A and PTEN genes located on chromosome 10q23.2 and 10q23.3 respectively, although others disagree 11).
In addition, generalized juvenile polyposis and juvenile polyposis coli (juvenile polyps restricted to the colorectum) have been defined 12). However, these forms appear to be variable expressions of the same disease, because patients of both forms have been reported to inherited according to a dominant mode in the same family 13). These forms may be sporadic, i.e., ‘de novo’, or inherited, and usually present later in childhood or in adult life. They are characterized by the presence of gastrointestinal juvenile polyposis and an increased risk of gastrointestinal cancer 14). A variety of extra-intestinal manifestations have been reported in these patients 15). In approximately 50% of juvenile polyposis coli or generalized juvenile polyposis syndrome cases, a heterozygous germline mutation in the SMAD4 or BMPR1A gene is identified 16). Several differences in phenotypic expression between carriers of a SMAD4 and BMPR1A mutations have been noted. SMAD4 mutations are associated with a more aggressive gastrointestinal phenotype, involving higher incidence of colonic adenomas and carcinomas and more frequent upper gastrointestinal polyps and gastric cancer than patients with a BMPR1A mutation 17). Also, the combined syndrome of juvenile polyposis syndrome and hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome) is associated with germline mutations in SMAD4 18).
Juvenile polyposis syndrome diagnosis
The diagnosis of juvenile polyposis syndrome relies primarily on the presence of certain clinical findings, including hamartomatous intestinal polyps (non-cancerous tissue masses) and/or a family history of juvenile polyposis syndrome.
Juvenile polyposis syndrome is clinically diagnosed if any one of the three following findings is present 19):
- More than five juvenile polyps in the colorectum
- Multiple juvenile polyps throughout the gastrointestinal tract
- Any number of juvenile polyps and a family history of juvenile polyps
In individuals with juvenile polyposis syndrome, juvenile polyps mainly involve the colon, but can also be seen in the stomach, small intestine and rectum. This type of polyp tends to retain mucous and is inflamed, which causes bleeding when the stool passes by the polyp and some of the surface cells shed.
Juvenile polyps can develop in infancy and into adulthood, but most individuals with juvenile polyposis syndrome will have polyps by the age of 20. Some individuals with juvenile polyposis syndrome may only have four or five polyps throughout their lifetime, others can have more than 100. The polyps associated with juvenile polyposis syndrome are most often benign; however, they can change into a malignant cancer.
A careful and detailed review of an individual’s medical and family history is important in diagnosing juvenile polyposis syndrome. A doctor or genetic counselor may construct a pedigree, or a multi-generation family tree, that indicates:
- Which members of the family have developed cancer
- The type of cancer developed
- The age of cancer onset
- The presence of any clinical manifestations
If the pattern of clinical features and/or cancers is suggestive of juvenile polyposis syndrome, the physician or counselor may recommend genetic testing be performed.
Genetic testing for juvenile polyposis syndrome
In order to confirm — on a molecular level — that an individual has juvenile polyposis syndrome, he or she can undergo the process of genetic testing which includes:
- A blood or saliva sample is obtained from an affected individual.
- DNA is isolated from the sample and the two copies of the both the BMPR1A and SMAD4 genes are evaluated using a variety of methods and compared to the normal reference sequences for BMPR1A and SMAD4.
- If a mutation in either BMPR1A or SMAD4 is identified, the genetic counselor can next examine whether the alteration has been previously reported in other individuals with juvenile polyposis syndrome.
About 40 percent of patients diagnosed with juvenile polyposis syndrome will have a mutation in either of these two genes (20 percent will have mutations in BMPR1A and 20 percent will have mutations in SMAD4).
However, it is important to remember that not all patients with juvenile polyposis syndrome carry a detectable alteration in BMPR1A or SMAD4. There are likely to be additional, undiscovered genes that play a role in the development of juvenile polyposis syndrome for the remaining 60 percent of patients. Therefore, the failure to identify an alteration in the BMPR1A or SMAD4 genes does not exclude the diagnosis of juvenile polyposis syndrome.
BMPR1A and SMAD4 genetic test results can also provide important information for other family members. Knowing the specific alteration that is present in an individual with juvenile polyposis syndrome allows other family members to undergo testing to determine whether they also carry the alteration and could therefore develop the features of juvenile polyposis syndrome.
Juvenile polyposis syndrome treatment
Clinical management of patients with symptomatic juvenile polyposis syndrome depends primarily on the anatomy of the gastrointestinal tract involved and polyp burden. Juvenile polyposis syndrome management involves routine colonoscopy with endoscopic polypectomy to reduce the risk of cancer, bleeding, and intestinal obstruction. When a large number of polyps are present, removal of all or part of the colon or stomach may be necessary.
If left untreated, polyps can cause bleeding and anemia (lowering of the levels of red blood cells). Routine colonoscopy and removal of the polyps reduces the risk of bleeding, blockage of the intestines, and/or development of colon cancer. Individuals with a large number of polyps may require removal of all or part of the colon or stomach, based on where the polyps are located.
The following surveillance recommendations are strongly recommended for anyone with a clinical diagnosis of juvenile polyposis syndrome or who has a family history of juvenile polyposis syndrome:
- Monitoring for rectal bleeding and/or anemia, abdominal pain, constipation, diarrhea or change in stool size, shape and/or color (any of these symptoms could warrant additional screening).
- Complete blood counts (CBC), colonoscopy, and upper endoscopy (procedure that allows for visualization of the inside lining of one’s digestive tract) beginning in the mid-teens (age 15) for baseline screening and repeated every 3 years into adulthood. These tests should be performed before age 15 if symptoms arise earlier.
- If only one or a few polyps are identified, the polyps should be removed. Subsequently, screening should be done annually until no additional polyps are found, at which time screening every 3 years may resume.
- If many polyps are identified, removal of most of the colon or stomach may be necessary. Subsequently, screening should be done annually until no additional polyps are found, at which time screening every 3 years may resume.
- Individuals with features of combined juvenile polyposis syndrome/hereditary hemorrhagic telangiectasia syndrome or with a known SMAD4 mutation can consider following hereditary hemorrhagic telangiectasia surveillance guidelines. Surveillance and treatment for features of hereditary hemorrhagic telangiectasia should be discussed with a physician with experience in the management of patients with hereditary hemorrhagic telangiectasia.
In addition to following recommended cancer surveillance guidelines, children and adults with juvenile polyposis syndrome should be encouraged to lead as healthy a lifestyle as possible, avoiding excess sun exposure and tobacco use. Patients and parents should be alert to signs of illness and pursue medical attention promptly should these occur.
There are no medicines to treat juvenile polyposis syndrome; however, COX 2 inhibitor therapy with sulindac may be tried for chemoprevention 20). Currently, nonsteroidal anti-inflammatory drugs (NSAID) chemoprevention in juvenile polyposis syndrome has not been systematically studied; however, two juvenile polyposis syndrome patients who had undergone proctocolectomy with pouch reconstruction and subsequent polypectomy from the pouch had no further polyp development in the pouch while on sulindac 21). However, the value of NSAID chemoprevention in juvenile polyposis syndrome requires further investigation.
Juvenile polyposis syndrome prognosis
Most juvenile polyps are benign; however, malignant transformation can occur. Lifetime estimated risk of developing gastrointestinal cancers in families with juvenile polyposis syndrome range from 9% to 50% 22) of individuals treated surgically and followed with surveillance. In a study by Aytac et al 23), 4/27 individuals with SMAD4 pathogenic variants and 0/8 individuals with BMPR1A pathogenic variants developed cancer. Most of the increased risk is attributed to colon cancer followed by cancers of the stomach, upper gastrointestinal tract, and pancreas. The incidence of colorectal cancer is 17%‐22% by age 35 years and approaches 68% by age 60 years 24). The median age at diagnosis is 42 years. In the study by Brosens et al 25), the relative risk for colorectal cancer was 34 in individuals with juvenile polyposis syndrome. The mean age of diagnosis of colorectal cancer was 43.9 years, with a cumulative lifetime risk of 38.7%. Therefore, due to the heightened risk of gastrointestinal malignancies in juvenile polyposis syndrome patients and families, a vigilant surveillance and robust management approach should be undertaken to mitigate the risks. The American College of Gastroenterology 26) recommends the following steps for the management and surveillance for juvenile polyposis syndrome patients:
- Surveillance of the gastrointestinal tract in affected or at‐risk juvenile polyposis syndrome patients should include screening for colon, stomach, and small bowel cancers.
- Colectomy and ileorectal anastomosis or proctocolectomy and ileal pouch anal anastomosis is indicated for polyp‐related symptoms, or when the polyps cannot be managed endoscopically.
- Cardiovascular examination for and evaluation for hereditary hemorrhagic telangiectasia should be considered for SMAD4 mutation carriers (conditional recommendation).
References [ + ]




{kind=link}