Contents
- Refractory epilepsy
- Refractory epilepsy causes
- Refractory epilepsy diagnosis
- Refractory epilepsy treatment
- Intractable epilepsy prognosis
Refractory epilepsy
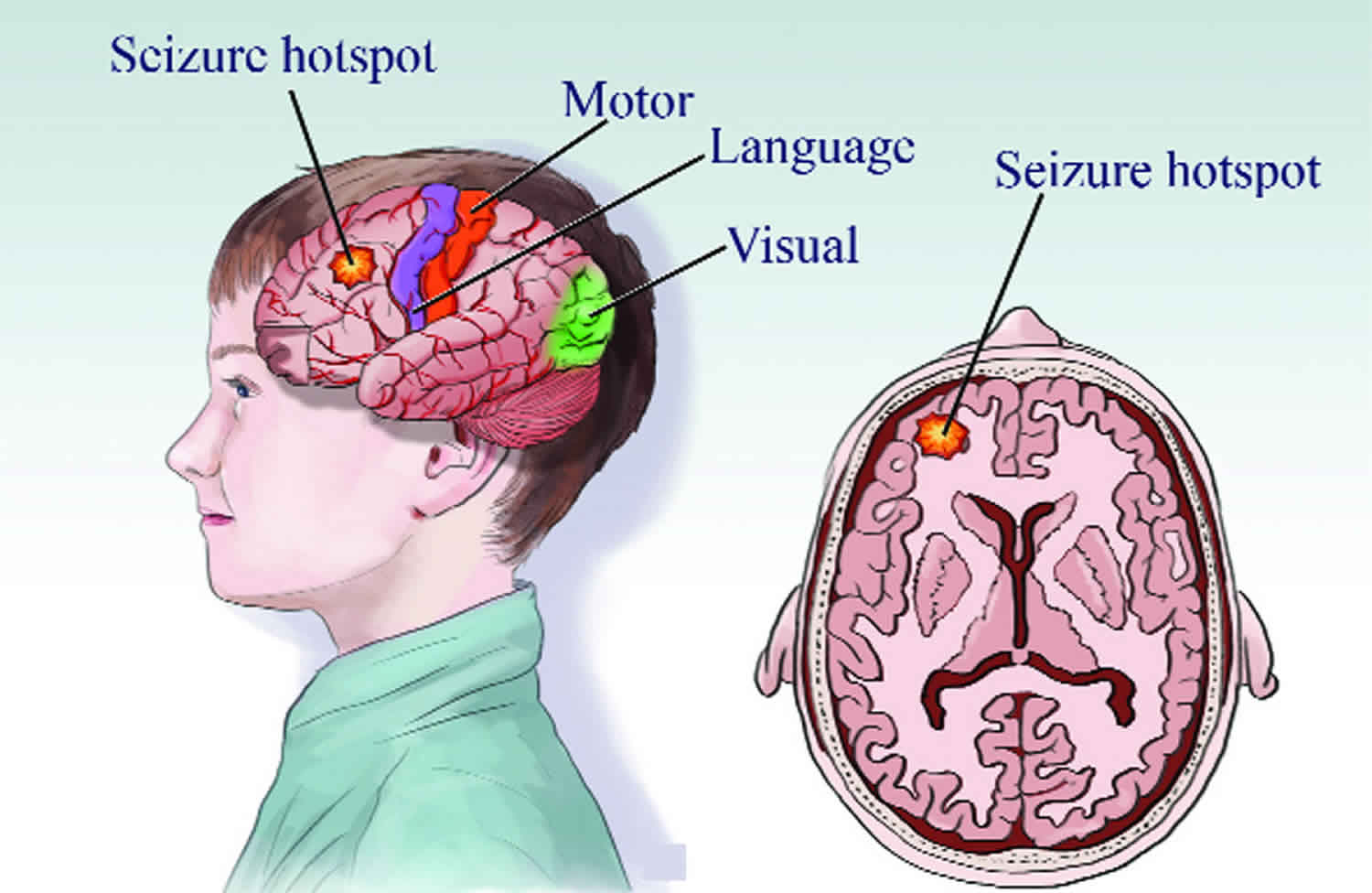
Intractable epilepsy also called “refractory epilepsy”, “uncontrolled epilepsy”, “difficult to control epilepsy” or “drug resistant epilepsy”, is hard to treat epilepsy that does not respond well to trials of at least two seizure medications 1). Some experts define a patient as having refractory seizures if treatment fails to achieve seizure freedom for 12 months or more, for whatever reason 2). The generally accepted definition of refractory seizures was proposed by Berg: failure of two or more drugs and occurrence of one or more seizures per month over 18 months 3).
The International League Against Epilepsy (https://www.ilae.org/files/dmfile/Epigraph-2016-2_Snead-9-MRE_Guidelines2016.pdf) has proposed the following definition of drug resistant epilepsy and suggests that this term be used instead of the term ‘refractory epilepsy’.
- Drug resistant epilepsy occurs when a person has failed to become (and stay) seizure free with adequate trials of two seizure medications called anti-epileptic drugs (AEDs).
- These seizure medications must have been chosen appropriately for the person’s seizure type, tolerated by the person, and tried alone or together with other seizure medications.
Studies suggest that epilepsy fails to come quickly under control with medicines in about one-third of cases, but the true frequency depends upon the definition of uncontrolled.
Most epilepsy specialists agree that refractory epilepsy is epilepsy for which seizures are frequent and severe enough, or the required therapy for them troublesome enough, to seriously interfere with quality of life 4).
Half of the people with epilepsy are children and up to 30% become refractory to medications and develop intractable epilepsy 5). Children with intractable epilepsy are exposed to many treatments and concomitant side effects 6) and have suboptimal growth and diets compared to healthy children 7).
However, in more recent years, the epilepsy community has recognized the need to continue striving for “no seizures” and the best control possible.
Seizures can be uncontrolled for four broad reasons.
- The diagnosis is wrong.
- The treatment is wrong.
- Despite the best treatment, triggers or lifestyle factors may affect seizure control.
- Properly diagnosed seizures do not respond to the best medical treatment.
Not all uncontrolled seizures are considered refractory or drug resistant. For example:
- If the diagnosis is corrected and seizures can be brought under control with a different treatment, then they would not be considered refractory.
- If triggers of lifestyle factors could be avoided or modified preventing breakthrough seizures, then medication therapy may work better. A person in this situation would not be considered drug resistant, but different drug trials may be considered and non-drug treatments may be considered to help control seizures.
Refractory epilepsy causes
Incorrect diagnosis
An incorrect diagnosis of epilepsy is more common than most people might think. One chart review study by Smith and colleagues in England found that 13% of patients referred for refractory epilepsy did not have epilepsy. If seizures are not controlled, then a reasonable first question is: “Are the episodes really seizures?” A number of conditions can imitate seizures. Some, but certainly not all, are listed here.
Imitators of epilepsy
- Fainting (syncope)
- Mini-strokes (transient ischemic attacks or TIAs)
- Hypoglycemia (low blood sugar)
- Migraine with confusion
- Sleep disorders, such as narcolepsy and others
- Movement disorders: tics, tremors, dystonia
- Fluctuating problems with body metabolism
- Panic attacks
- Nonepileptic (psychogenic) seizures
Experienced clinicians are skilled at using a combination of the medical history, the physical exam and certain laboratory tests to determine whether sudden episodes with alteration in sensation, strength, behavior or awareness are seizures or one of the imitators. But sometimes this is difficult. People have been referred to epilepsy centers for brain surgery, when their underlying condition was not epilepsy, but one of the imitators.
Poor or less than optimal seizures treatment
Another reason for uncontrolled seizures is poor or less than optimal treatment. In other words, the ‘wrong treatment’ is being used to treat the seizures. Common reasons for suboptimal treatment are listed below.
Reasons for suboptimal treatment of seizures:
- Using the wrong medication
- Inadequate doses of medicine
- Polypharmacy and toxicity
- Missing doses (poor compliance)
- Complicating factors (illness, sleep deprivations, extreme stress).
True intractable epilepsy or difficulty controlling seizures can result from not tolerating seizure medications or seizures not responding to the medicines.
All medications have potential side effects, but some people experience them more often than others, or the side effects are more bothersome. Sometimes people develop allergies to medicines or just can’t tolerate non- allergy side effects. People who are very sensitive to seizure medicines are less likely to find one that they can tolerate and that will work! Seizures that might be easy to treat with medicine become hard to treat when the best medicines are off-limits. Some people with multiple drug resistance have a type of metabolism that quickly inactivates or isolates drugs, causing them to be less effective. When this happens, exploring other treatments like surgery may be helpful.
Another common problem is reaching a “honeymoon” state or as it is officially known, developing medication “tolerance.” In this situation, a new drug works for a few months and then seizures return. The cycle repeats with each new medication. Such patients can end up on a stressful “rotation diet” of different medicines. It is another form of drug resistance.
When seizures persist after at least two good trials of the proper drugs at the right dose, a person would be considered to have intractable or drug resistant epilepsy.
Using the wrong medication
Many seizure medications have useful actions against a number of different seizure types. But some medicines are not right for certain types of seizures. Carbamazepine (Tegretol), for example is usually good for treating complex partial seizures, but not absence seizures. Ethosuximide (Zarontin) is good for absence, but not complex partial seizures. Since absence and complex partial seizures can occasionally be confused with each other, there is a chance for using the wrong medicine.
Inadequate or incorrect doses of medicine
People vary widely in their response to seizure medicines. Every medicine has a suggested dosage range, but that range is too high for some and too low for others. If a dose that is too high for an individual is used, a person will have too many side effects. A dose that is too low may lead to seizures.
- Some people with uncontrolled seizures may become seizure free when the medication daily dosages are increased.
- Others may do better on low doses of antiepileptic drugs (AEDs), which leads to less medication side effects.
- Measuring blood levels of antiepileptic drugs (AEDs) sometimes helps to guide therapy, but levels are not as important as carefully asking about side effects and seizure control. The newer seizure medicines often have fewer side effects than the older seizure medicines.
Polypharmacy and toxicity
Polypharmacy is the use of several medications at once to treat the same condition. Some people require more than one drug to control their epilepsy, but additional medications rarely lead to complete freedom from seizures.
Two important studies, one by Mattson and colleagues 8) and the other by Kwan and Brodie 9) suggest that if a person is not seizure-free on a good dosage of a single antiepileptic drug, then adding a second will make them seizure-free only about 10% of the time. The second drug may help, but not usually to the point of complete control. Two drugs have more side effects than does one drug, and three drugs more than two.
Patients taking polypharmacy may have so many side effects that it is often difficult for someone to tolerate a higher dose for any of their antiepileptic drugs.
Also, polypharmacy can lead to drug interactions that limit how well the drug may work or increases side effects of another drug.
One way to treat refractory seizures in people taking many medications is to streamline or simplify the medicines. Sometimes “less can be more,” especially if it lowers overall levels of side effects and allows an increase in the drug that is most effective. Making these changes can be hard, with a period of seizures and side effects during the changes, until the new and improved regimen is established.
Missing antiepileptic drug doses (poor adherence or compliance)
Missing medication is a cause of breakthrough seizures. Almost everyone forgets to take pills, especially if the pill schedule is complicated. In the medical field, this is called “poor compliance.” Learn about the importance of adherence and ways to make taking medications easier It can make a real difference!
Complicating factors (illness, sleep deprivations, extreme stress)
Complicating or precipitating factors for seizures can make them more difficult to control. These again vary with the individual. Triggers may include alcohol, exercise, flashing lights or certain patterns, general illness, heavy breathing (hyperventilation), lowering dose of medicines, taking certain medications, the menstrual cycle, missing medications, missing sleep, recreational drugs, and stress. All too often, a seizure breakthrough is preceded by one of these, or other personally relevant, factors.
Refractory epilepsy diagnosis
Diagnosing epilepsy is not simple. Doctors gather lots of different information to assess the causes of seizures. If you have had two or more seizures that started in the brain you may be diagnosed with epilepsy. Getting a diagnosis is not always easy as there is no single test that can diagnose epilepsy. If there is a possibility that you have epilepsy, the National Institute for Health and Care Excellence recommends that you are referred to an epileptologist (a doctor who is trained in diagnosing and treating epilepsy) within two weeks.
Your diagnosis is based on finding out what happened to you before, during and after your seizures. For example, some types of faints can look like epileptic seizures, and often before fainting a person feels cold, clammy and their vision goes blurry. But epileptic seizures happen very suddenly and a person may have no warning that a seizure is about to happen.
A number of tests are used to determine whether a person has a form of epilepsy and, if so, what kind of seizures the person has.
Medical history
Taking a detailed medical history, including symptoms and duration of the seizures, is still one of the best methods available to determine what kind of seizures a person has had and to determine any form of epilepsy. The medical history should include details about any past illnesses or other symptoms a person may have had, as well as any family history of seizures. Since people who have suffered a seizure often do not remember what happened, caregiver or other accounts of seizures are vital to this evaluation. The person who experienced the seizure is asked about any warning experiences. The observers will be asked to provide a detailed description of events in the timeline they occurred.
Blood tests
Blood samples may be taken to screen for metabolic or genetic disorders that may be associated with the seizures. They also may be used to check for underlying health conditions such as infections, lead poisoning, anemia, and diabetes that may be causing or triggering the seizures. In the emergency department it is standard procedure to screen for exposure to recreational drugs in anyone with a first seizure.
Imaging and monitoring
An electroencephalogram, or EEG, can assess whether there are any detectable abnormalities in the person’s brain waves and may help to determine if antiseizure drugs would be of benefit. This most common diagnostic test for epilepsy records electrical activity detected by electrodes placed on the scalp. Some people who are diagnosed with a specific syndrome may have abnormalities in brain activity, even when they are not experiencing a seizure. However, some people continue to show normal electrical activity patterns even after they have experienced a seizure. These occur if the abnormal activity is generated deep in the brain where the EEG is unable to detect it. Many people who do not have epilepsy also show some unusual brain activity on an EEG. Whenever possible, an EEG should be performed within 24 hours of an individual’s first seizure. Ideally, EEGs should be performed while the person is drowsy as well as when he or she is awake because brain activity during sleep and drowsiness is often more revealing of activity resembling epilepsy. Video monitoring may be used in conjunction with EEG to determine the nature of a person’s seizures and to rule out other disorders such as psychogenic non-epileptic seizures, cardiac arrhythmia, or narcolepsy that may look like epilepsy.
A magnetoencephalogram (MEG) detects the magnetic signals generated by neurons to help detect surface abnormalities in brain activity. MEG can be used in planning a surgical strategy to remove focal areas involved in seizures while minimizing interference with brain function.
The most commonly used brain scans include CT (computed tomography), PET (positron emission tomography) and MRI (magnetic resonance imaging). CT and MRI scans reveal structural abnormalities of the brain such as tumors and cysts, which may cause seizures. A type of MRI called functional MRI (fMRI) can be used to localize normal brain activity and detect abnormalities in functioning. SPECT (single photon emission computed tomography) is sometimes used to locate seizure foci in the brain. A modification of SPECT, called ictal SPECT, can be very helpful in localizing the brain area generating seizures. In a person admitted to the hospital for epilepsy monitoring, the SPECT blood flow tracer is injected within 30 seconds of a seizure, then the images of brain blood flow at the time of the seizure are compared with blood flow images taken in between seizures. The seizure onset area shows a high blood flow region on the scan. PET scans can be used to identify brain regions with lower than normal metabolism, a feature of the epileptic focus after the seizure has stopped.
Developmental, neurological, and behavioral tests
Tests devised to measure motor abilities, behavior, and intellectual ability are often used as a way to determine how epilepsy is affecting an individual. These tests also can provide clues about what kind of epilepsy the person has.
Refractory epilepsy treatment
When seizures are not controlled, despite best efforts, different therapies are available to people with epilepsy and their health care team.
- Talk to your doctor to reevaluate the diagnosis and medication therapy.
- Make sure you are doing everything you can to take the medicines consistently and manage or avoid seizure triggers.
- Consider a referral to an epilepsy specialist called an epileptologist and specialized epilepsy center. An Epilepsy Center takes a comprehensive approach to medical, psychological, and social problems associated with seizures. They should be capable of providing the full range of treatments for epilepsy, both medication and non-drug therapies.
- Find an American Epilepsy Society Doctor (https://my.aesnet.org/FindaDoctor)
- Find an Epilepsy center (https://www.naec-epilepsy.org/about-epilepsy-centers/find-an-epilepsy-center)
- Learn about non-drug therapies for epilepsy
- Epilepsy surgery
- Neurostimulation devices
- Dietary therapy
- Experimental trials
Antiepileptic drug therapy
There are at least 21 antiepileptic drugs available today, 15 of which have been recently released. Given that there are few robust randomized controlled trials or comparative studies, determining which antiepileptic drug to use can be challenging. The following general principles can be applied. If the trial of two antiepileptic drugs is found to be ineffective by a neurologist, patients should be referred to an epileptologist as per the Provincial Guidelines for the Management of Epilepsy in Adults and Children 10).
Practice recommendations for antiepileptic drug trial:
- Optimize the dose of each antiepileptic drug by increasing the dose incrementally. If the maximum dose is ineffective, introduce a second antiepileptic drug while continuing on the first. If seizure control is achieved, consider tapering the first antiepileptic drug. The advice to “start low and go slow” is appropriate 11).
- If one or two antiepileptic drugs are ineffective, rational polytherapy should be explored. There is little systematic study of rational polytherapy. Considerations include a higher incidence of side effects when multiple antiepileptic drugs are used.
- Consider using antiepileptic drugs with different mechanisms of action. However, although mechanisms of action have been described for a number of antiepileptic drugs, it is not certain that these are their only mechanisms of action or even the most important. For example, Levetiracetam affects the SV2a receptor on the synaptic vesicle but also has calcium channel modulating effects and GABAergic properties.
- Avoid using an antiepileptic drug that may worsen or provoke seizures. Carbamazepine, Oxcarbazepine, Phenytoin, Vigabatrin and Tiagabine may worsen myoclonus and absence seizures. Gabapentin and Lamotrigine may worsen myoclonus 12). Benzodiazepines given intravenously may worsen tonic seizures but may be very useful in treating Lennox-Gastaut and does not contraindicate their use 13).
Recent literature has concentrated on the most recently introduced antiepileptic drugs. There have been few class 1 studies and no comparative studies done on these new antiepileptic drugs 14). The most recently available antiepileptic drugs are Rufinamide, Lacosamide, Perampanel, Eslicarbazepine and Retigabine (ezogabine). As per literature its use is limited because of the long term effect of developing a blue hue to the skin and retina 15).
Despite the agreed need for consideration of many factors in deciding which antiepileptic drug to use, the most important consideration is still the type of seizure. Greater precision can be applied when a particular epilepsy syndrome is identified (e.g. childhood absence epilepsy, juvenile myoclonic epilepsy) but many patients do not have an easily identifiable syndrome. Using a broad-spectrum anticonvulsant may be an efficient approach for the large number of children who do not have a defined epilepsy syndrome. These broad spectrum anticonvulsants are:
- Valproate
- Levetiracetam
- Lamotrigine
- Topiramate
- Clobazam
Clobazam is safe and effective for seizures associated with Lennox-Gastaut syndrome 16), focal epilepsy in tuberous sclerosis complex and other refractory epilepsies in childhood. There is no data to recommend one of these antiepileptic drugs over another and the age of patient, concomitant medications, potential side effects, ease of use and cost to the patient are important considerations when data on efficacy and effectiveness are lacking.
Ideal antiepileptic drug polytherapy would combine supra-additive (synergistic) efficacy with infra-additive toxicity. The following list of anticonvulsants classified by mechanism of action is intended to encourage the use of antiepileptic drugs with different mechanisms of action rather than combining antiepileptic drugs with the same mechanism of action.
Limited data has suggested the following combinations 17).
- Ethosuximide-Valproate
- Lacosamide-Levetiracetam
- Stiripentol-Clobazam
- Lamotrigine-Valproate
NOTE: This combination has the best human evidence for synergy especially for focal seizures. The question whether the addition of valproate causes apparent synergy by inhibiting lamotrigine metabolism and increasing lamotrigine levels has been studied and the limited data does not show this. Nonetheless, because of the recognised effect of valproate on lamotrigine, the latter drug should be introduced very cautiously in patients on valproate. Current practice would be to use doses of lamotrigine that are 25% of usual introductory doses and in children a maximum dose of 5 mg/kg/day is suggested. However, the introduction of valproate in someone who is already on lamotrigine is said not to cause any risk for sensitivity reactions such as Stevens-Johnson syndrome or toxic epidermal necrolysis.
For the common medical refractory childhood epilepsy syndromes the following suggestions are made 18):
- Infantile spasms: ACTH, high dose prednisone/prednisolone, vigabatrin,
- Lennox-Gastaut Syndrome: Lamotrigine, topiramate. Lamotrigine can exacerbate myoclonic seizures in select patients. Lamotrigine and topiramate appear to have a synergistic effect. Rufinamide and clobazam also have demonstrated effect.
- Severe Myoclonic Epilepsy of Infancy (Dravet syndrome): Because this is a SCN1A-based voltage gated sodium channel disorder, antiepileptic drugs targeting this should not be used. Recommended treatments are valproate, clonazepam and clobazam. The addition of stiripentol to valproate and clobazam has been shown to be effective
- Landau-Kleffner Syndrome or Electrical Status Epilepticus in Sleep: High dose roal diazepam at night, valproate, levetiracetam. If antiepileptic drugs are ineffective, immunomodulatory treatments should be considered including steroids and intravenous immune globulin alone (IVIg).
Vagus nerve stimulation
Vagus nerve stimulation therapy, or vagus nerve stimulation, is a way of controlling seizures in people who do not respond to medications and may not respond to surgery. The vagus nerve sends information from your neck down to the chest and stomach, and then back up again. The vagus nerve then sends information up to the brain. Stimulation of the vagus nerve can change the likelihood of the brain to have seizures.
Vagus nerve stimulation therapy consists of a device placed under the skin in the left side of the chest. An electrode attached to the generator is then placed under the skin and connects with the vagus nerve in the left side of the neck.
Preprogrammed stimulation can be delivered from the generator in the chest to the vagus nerve. Settings can be adjusted by a nurse and doctor trained in the use of vagus nerve stimulation therapy.
The stimulation doesn’t work right away, but after a few months of therapy, about 25 to 30% of people may see that seizures decrease by 50% or more. Usually seizure control improves over time, with up to about 45% of people having seizures decrease at least by 50% after 1 to 2 years of therapy.
- Complete seizure freedom by vagus nerve stimulation happens in only small numbers of people. And in some people, it doesn’t work at all.
- Vagus nerve stimulation is not considered a substitute for seizure medications. People continue to take seizure medications while using vagus nerve stimulation. However, if the vagus nerve stimulation works, some people can lower the number or dose of medications and lessen side effects.
- Side effects of vagus nerve stimulation are usually mild, including hoarseness and coughing, mostly while becoming use to the stimulation.
- Vagus nerve stimulation is also approved by the U.S. Food and Drug Administration (FDA) for depression that does not respond to other treatments.
Dietary therapies
When medicines don’t work, dietary therapies have been found to help in a number of people. Like surgery or vagus nerve stimulation, it doesn’t work in everyone. It has been used most often in children, as it’s easier to control what young children eat. However, some of the diets have also been used in adults and showed very promising results. The diets used most often include:
- Ketogenic Diet
- Modified Atkins Diet
- Low Glycemic Index Treatment
Ketogenic diet
The “classic” ketogenic diet is a special high-fat, low-carbohydrate diet that helps to control seizures in some people with epilepsy. The name ketogenic means that it produces ketones in the body (keto = ketone; genic = producing). Ketogenic diet is prescribed by a physician and carefully monitored by a dietitian. It is usually used in children with seizures that do not respond to medications. It is stricter than the modified Atkins diet, requiring careful measurements of calories, fluids, and proteins. Foods are weighed and measured.
Ketones are formed when the body uses fat for its source of energy. Usually the body uses carbohydrates (such as sugar, bread, pasta) for its fuel. Because the ketogenic diet is very low in carbohydrates, fats become the primary fuel instead. The body can work very well on ketones (and fats).
Ketones are not dangerous. They can be detected in the urine, blood, and breath. Ketones are one of the more likely mechanisms of action of the diet, with higher ketone levels often leading to improved seizure control. However, there are many other theories for why the diet will work.
The ketogenic diet is a treatment for intractable epilepsy and typically a third of the patients will experience a greater than 90% reduction in seizure frequency 19). The ketogenic diet is often discontinued after three years, but in children whose seizures return during a gradual taper, the ketogenic diet can be extended for more than 10 years 20). Ketogenic diet treatment can result in height velocity deceleration and growth failure 21).
Children who are on the ketogenic diet continue to take seizure medicines. Some are able to take smaller doses or fewer medicines than before they started the diet. This is usually attempted after 1 month on the diet. If the person goes off the ketogenic diet for even one meal, it may lose its good effect. So it is very important to stick with the ketogenic diet as prescribed. Being on a ketogenic diet allows for a sense of control over seizures by a parent (or person living with epilepsy). Many families comment on this and the how their ability to cook helps their child. Most major pediatric hospitals (and countries) have ketogenic diet centers – just ask.
Are any other medicine changes needed?
Because the ketogenic diet does not provide all the vitamins and minerals found in a balanced diet, the dietician will recommend vitamin and mineral supplements. The most important of these are calcium and vitamin D (to prevent thinning of the bones), B Vitamins, and selenium.
There are no anticonvulsants (seizure medicines) that should be stopped while on the ketogenic diet. Topiramate (Topamax) and zonisamide (Zonegran) do not have a higher risk of kidney stones while on the diet, but zonisamide can increase the chance of stones.
Medication levels do not likely change while on the ketogenic diet according to recent studies. All medications should be in as carbohydrate/sugar-free a form as possible to avoid hidden sugars. Most liquids are changed by pharmacists to pills.
Who will ketogenic diet help?
- Doctors usually recommend the ketogenic diet for children whose seizures have not responded to several different seizure medicines.
- The classic ketogenic diet diet is usually not recommended for adults, mostly because the restricted food choices make it hard to follow. However, the modified Atkins diet does work well. This also should be done with a good team of adult neurologists and dietitians.
- The ketogenic diet has been shown in many studies to be particularly helpful for some epilepsy conditions. These include infantile spasms, Rett syndrome, tuberous sclerosis complex, Dravet syndrome, Doose syndrome, and GLUT-1 deficiency. Using a formula-only ketogenic diet for infants and gastrostomy-tube fed children may lead to better compliance and possibly even improved efficacy.
- Recent studies have also shown that infants can be successfully started on dietary therapy too (https://www.epilepsy.com/article/2017/1/ketogenic-diet-neonates).
- Learn about Ketogenic Guidelines for Infants (https://www.epilepsy.com/article/2016/8/ketogenic-diet-guidelines-infants).
- The ketogenic diet works well for children with focal seizures, but may be less likely to lead to an immediate seizure-free result.
- In general, the ketogenic diet can always be considered as long as there are no clear metabolic or mitochondrial reasons not to use it.
- The ketogenic diet is sometimes started to help reduce or even stop anti-seizure drugs. However, that does not always occur – often it is a “partnership” between drugs and food to help reduce seizures that works.
What is it like to be on the ketogenic diet?
The typical “classical” ketogenic diet, called the “long-chain triglyceride diet,” provides 3 to 4 grams of fat for every 1 gram of carbohydrate and protein. That is about 90% of calories from fat.
Usually when the classic ketogenic diet is prescribed, the total calories are matched to the number of calories the person needs. For example, if a child is eating a 1500 calorie regular diet, it would be changed to a 1500 calorie ketogenic diet. For very young children only, the diet may be prescribed based on weight, for example 75 to 100 calories for each kilogram (2.2 pounds) of body weight. If it sounds complicated, it is. That’s why people need a dietician’s help when using ketogenic diet.
A ketogenic diet “ratio” is the ratio of fat to carbohydrate and protein grams combined.
- A 4:1 ratio is more strict than a 3:1 ratio and is typically used for most children.
- A 3:1 ratio is typically used for infants, adolescents, and children who require higher amounts of protein or carbohydrate for some other reason.
The kinds of foods that provide fat for the ketogenic diet are butter, heavy whipping cream, mayonnaise, and oils (e.g., canola or olive). Because the amount of carbohydrate and protein in the diet have to be restricted, it is very important to prepare meals carefully. No other sources of carbohydrates can be eaten.
The ketogenic diet is supervised by:
- a dietician who monitors the child’s nutrition and can teach parents and the child what can and cannot be eaten
- a neurologist who monitors medications and overall benefits
What happens first?
Typically the ketogenic diet is started in the hospital. The child traditionally begins by fasting (except for water) under close medical supervision for 18-24 hours. The ketogenic diet is then started, either by slowly increasing the calories or the ratio.
There is growing evidence that fasting is probably not necessary for long-term efficacy, although it does lead to a quicker onset of ketosis. Most centers today do NOT start with a fasting period. The primary reason for admission in most centers is to monitor for any increase in seizures on the diet, ensure all medications are carbohydrate-free, and educate the families.
Does ketogenic diet work?
Several studies 22), 23) have shown that the ketogenic diet does reduce or prevent seizures in many children whose seizures could not be controlled by medications.
- Over half of children who go on the diet have at least a 50% reduction in the number of their seizures.
- Some children, usually 10-15%, even become seizure-free.
Ketogenic diet side effects
- A person starting the ketogenic diet may feel sluggish for a few days after the diet is started. This can worsen if a child is sick at the same time as the diet is started.
- Make sure to encourage carbohydrate-free fluids during illnesses.
- Other side effects that might occur if the person stays on the diet for a long time are:
- Kidney stones
- High cholesterol levels in the blood
- Constipation
- Slowed growth
- Bone fractures
How is the patient monitored over time?
- Early on, the doctor will usually see the child every 1 to 3 months.
- Blood and urine tests are performed to make sure there are no medical problems.
- The height and weight are measured to see if growth has slowed down.
Can the ketogenic diet ever be stopped?
If seizures have been well controlled for some time, usually 2 years, the doctor might suggest going off the ketogenic diet.
- Usually, the person is gradually taken off the ketogenic diet over several months or even longer. Seizures may worsen if the ketogenic diet is stopped all at once.
- Children usually continue to take seizure medicines after they go off the ketogenic diet.
- In many situations, the ketogenic diet has led to significant, but not total, seizure control. Families may choose to remain on the ketogenic diet for many years in these situations.
Responsive neurostimulation
At least 30% of people do not respond to seizure medicines. Some people can have surgery to remove where seizures start in the brain. This treatment is the only way to cure epilepsy, but it doesn’t work in everyone. On average, only about 60% of people can be free of disabling seizures from removal of a seizure focus in the temporal lobe. Vagus nerve stimulation therapy or dietary therapies, such as the ketogenic diet, may help many people. Another option is responsive neurostimulation. Responsive Neurostimulation is also known as as RNS® Therapy, is a new seizure treatment was approved by the U.S. Food and Drug Administration (FDA) in 2013.
The RNS® System is similar to a heart pacemaker. It can monitor brain waves, then respond to activity that is different from usual or that looks like a seizure. People cannot feel the stimulation once it’s programmed. It doesn’t cause pain or any unusual feelings. Responsive Neurostimulation Therapy has shown to reduce seizures and improve quality of life in most people who have used it.
Everyone’s seizures are a bit different, either in type, number, or pattern. Therefore an ideal way to treat seizures is personalizing the treatment to each person. The ability to give the treatment only when it’s needed (at the time of a seizure or suspected seizure activity in the brain) is a key feature of the RNS® System.
The Responsive Neurostimulation System is designed to work in 3 key ways:
- Monitor brain waves at the seizure focus, all the time – even during sleep.
- Detect unusual electrical activity that can lead to a seizure.
- Respond quickly (within milliseconds) to seizure activity by giving small bursts or pulses of stimulation. This goal is to help brainwaves return to normal, even before it could turn into a seizure.
Most comprehensive epilepsy centers that provide epilepsy surgery can also offer the Responsive Neurostimulation System. Before having the RNS placed, a person must go though detailed testing to see where their seizures arise in the brain.
- The RNS® System is similar to a heart pacemaker. It can monitor brain waves, then respond to activity that is different from usual or that looks like a seizure.
- A device or stimulator is placed in the bone covering the brain. Tiny wires or leads are placed in one or two places on top of the brain where seizure activity may begin. These wires connect to the stimulator. Once the wires and device are placed, nothing can be seen.
- The system can give small pulses or bursts of stimulation to the brain when anything unusual is detected. This can stop seizure activity before the actual seizure begins. Or it could stop seizure activity from spreading from a small focal seizure to a generalized seizure.
- People cannot feel the stimulation once it’s programmed. It doesn’t cause pain or any unusual feelings.
- It’s not permanent. It can be turned off or removed if it doesn’t work or a person doesn’t wish to use it any longer.
Who can use the Responsive Neurostimulation System?
- The RNS® System has been approved by the FDA to treat focal or partial seizures in adults, 18 years and older.
- It’s used in addition to seizure medictions. This is called adjunctive or add-on treatment.
- It’s designed for people with refractory seizures. This means that a person continues to have seizures despite at least trials with two seizure medications.
- It’s used in people who can not have epilepsy surgery to remove where the seizures start or resective surgery has not worked.
How does the Responsive Neurostimulation System work?
A device like the Responsive Neurostimulation system changes or modulates brain activity to stop or prevent seizures.
The exact way that the Responsive Neurostimulation System works is not known. It is thought to act on a certain substance in the brain called an inhibitory neurostramitter. This type of substance acts to inhibit or stop activity from brain cells that could lead to seizures. This may explain how the RNS works in the short term. Yet it’s long-term effect may be caused by something else affecting how brain cells work more broadly.
How helpful is Responsive Neurostimulation Therapy?
Although the Responsive Neurostimulation System is not a cure for epilepsy, it has shown to reduce seizures in most people who have used it. So far, these effects appear to improve over time in many people.
Seizure Reducton Results
- 230 patients with the RNS® System were followed over time in a controlled trial 24). The average decrease in seizures was 44% after 1 year, 53% at 2 years, and up to 66% after 3 to 6 years of using Responsive Neurostimulation.
- The same trend was seen when some of these people were followed for 7 years. Seizures decreased by an average of 72%.
- So far, 2 out of 3 people with the Responsive Neurostimulation System (66%) had their seizures cut in half after 7 years of using it.
- Some people had extended times of being seizure free as well. In the open label study or long-term study, 1 out of 3 people reported periods of no seizures for 6 months in a row.
Quality of Life Results
Benefits of any therapy must also look at how a person feels and their quality of life. A study of 191 people 25) with the Responsive Neurostimulation System found improvements in quality of life aside from seizure control. These benefits did not appear due to changes in seizures or medicines. These included:
- Physical health
- Cogntitive functioning (for example thinking, remembering, and concentrating)
- Emotional health or mood
- Less worry about seizures
- Overall quality of life
Epilepsy surgery
- Surgery is a treatment option that offers some people a real solution to stopping their seizures.
- Surgery works best for people who have seizures that always begin in one area of the brain.
- There are many different types of epilepsy surgery. The type of surgery recommended will depend on many factors. The key factors are the type of seizures and where they start in the brain.
- Surgery typically involves opening the skull and removing the area of the brain where seizures start. There are less invasive options available too.
- Ask your health care team whether epilepsy surgery is an option for you.
Epilepsy surgery is a treatment used to help control seizures when seizures are not controlled by medications.
Epilepsy surgery is “neurosurgery,” which means it is surgery involving the brain. Doctors who do epilepsy surgery are neurosurgeons. Neurosurgeons who specialize in the care of people with seizures are found at comprehensive epilepsy centers.
Epilepsy centers are typically multi-disciplinary, meaning they have a coordinated team of experts in adult and pediatric epilepsy care, including:
- Epileptologists (neurologists who specialize in epilepsy)
- Nurses
- Psychologists
- Neurosurgeons who specialize in epilepsy
- Neuroradiologists who specialize in brain imaging
With more than one epileptologist on staff, there is a team of experts available to discuss challenging cases. The availability of broad expertise and input from colleagues in the same specialty is not available in a single-epileptologist clinic.
A comprehensive epilepsy center:
- Permits experts in multiple areas or disciplines to engage the person living with epilepsy and their caregivers
- Maximizes interaction between colleagues with an interest in epilepsy
- Facilitates coordination of care of people living with epilepsy and their families
Epilepsy surgery key points:
- Epilepsy surgery works best for people who have seizures that always begin in one area of the brain.
- Tests are done before surgery. This testing helps to find the area in the brain where seizures start. The tests also help to make sure the area where seizures begin is not involved in important brain functions that include speech, movement, or memory.
- Epilepsy surgery can reduce the number and severity of seizures a person has when compared to only taking medications.
- By removing the seizure focus in the brain, epilepsy surgery can, in most cases, successfully and safely stop seizures. Some people may need a second surgery to become seizure free. Although many people are permanently seizure free after surgery, seizures can come back in some people.
What actually happens during epilepsy surgery?
Currently, most cases of epilepsy surgery involve removing the area where seizures start. Typically, this involves creating an opening in the skull (the bone covering the brain) in a procedure called a craniotomy.
- Epilepsy surgery usually takes several hours.
- The person’s hair may be clipped short or the head shaved in the area where surgery will be done. This lessens the risk of infection.
- Anesthesia is given so the person is asleep and not aware during the surgery.
- Just like any surgery, heart rate, blood pressure, and oxygen levels are checked closely.
- A small part of the skull is opened so the surgeon can see the area of abnormal tissue to be removed.
- EEG (electroencephalogram) monitoring may be done during the surgery. This will confirm exactly where in the brain the seizures start and what area needs to be removed.
- In some cases, the person is woken up during the surgery so he or she can talk and respond to questions. This helps the surgeon test which areas of the brain control speech or movement. This is important to lessen the risks of these areas being affected during surgery. This part of the surgery is called “brain mapping.”
- Once the brain areas are mapped, the anesthesiologist will return the person to sleep with medication, and the neurosurgeon can safely remove the abnormal brain tissue causing the seizures.
- The piece of bone that was removed from the skull at the beginning of the surgery will then be secured back in its normal position. The skin overlying the skull is then closed, and a head bandage is applied.
What happens after epilepsy surgery?
Right after surgery, the person will be in a special recovery unit where the doctors and nurses will monitor you very closely as you wake up from anesthesia. Depending on the hospital, a person may spend the first night after surgery in the intensive care unit for close monitoring or may go directly to the neurosurgery or epilepsy unit.
In the first week after surgery:
- Some swelling of the scalp and face is usually seen. This is normal as the tissue will need some time to heal from the surgery.
- Headaches are also common.
- Medicines to treat these symptoms will be given.
- For most people, the swelling and head pain after surgery goes away within a few weeks.
Typically, people spend 3 to 4 days in the hospital after surgery. In some cases, a longer stay in the hospital is needed.
Usually, people can go home to recover. If a person needs more help or lives alone, they may go to a rehabilitation facility for a short time until they are ready to go home.
A person recovering from surgery needs to rest and slowly return to their normal daily routines:
- Activity is gradually increased.
- Most people are back to their usual activities in 4 to 6 weeks.
- There may be some restrictions or precautions depending on each person’s situation.
Seizure medicines are continued after surgery. Medicine helps to protect the brain during the healing and increases the person’s chances of being seizure free later on.
- If a person is seizure free after a year or more, medicines may be gradually lowered or tapered off.
- Changing medicines slowly may help the person’s chances of staying seizure free.
Each person should talk with their epilepsy team about what is best in their situation.
There are other steps to consider in recovering from brain surgery.
- One of the goals of epilepsy surgery is to improve a person’s quality of life. This may include support from a therapist, vocational counselor, social worker, physical or occupational therapist, or neuropsychologist.
- Finding support as you adjust to changes after surgery is an important part of your recovery.
- Remember, your epilepsy team will be there to support you through all stages leading up to surgery, during surgery, and recovery.
Immunotherapy
The main treatment options other than antiepileptic drugs include medications used when the immune system is involved. Evidence that the immune system is involved in the pathogenesis of epilepsy particularly, medically refractory epilepsy, has given rise to the use of adjunctive immunotherapy to slow or change the epileptogenic process. Medications include immunoglobulins, corticosteroids, plamapharesis and monocloncal antibodies such as rituximab, natalizumab. There is limited data of these treatments outside of specific epileptic encephalopathies such as West syndrome, Rasmussen encephalitis, Landau Kleffner and specific anitbody mediated encephalitis such as anti NMDA encephalitis.
Corticosteroids form one of the main treatment options. Corticosteroids cause immunosuppression by decreasing the function and numbers of lymphocytes, including both B cells and T cells. By inhibiting a critical transcription factor involved in the synthesis of many mediators (i.e., cytokines) and proteins (i.e., adhesion proteins) that promote an immune response, they blunt the capacity of the immune system to mount a response.
Corticosteroids have an anti-inflammatory effect by preventing the formation of prostaglandins and leukotrienes, two main factors in inflammation. This is mediated by the release of lipocortin which by inhibition of phospholipase A2 reduces arachidonic acid release.
Corticosteroids have been used as therapy in many epileptic syndromes including infantile spasms, an age specific epilepsy syndrome associated with epileptic spasms, and in many cases with neurodevelopmental regression and an EEG finding of hypsarrhythmia (West Syndrome).
Low dose ACTH, or vigabatrin should be considered for the treatment of infantile spasms. However hormonal therapy with prednisolone or other steroids have also been used but the review by Go et al. 26) found there was little evidence to suggest that prednisolone, dexamethasone, and methylprednisolone are as effective as ACTH for short-term treatment of infantile spasms.
Steroids are also used in Rasmussen’s Encephalitis which is a rare, sporadic but potentially severe immune-mediated brain disorder leading to unilateral hemispheric atrophy, associated progressive neurological dysfunction and poorly controlled seizures.
Prednisolone or prednisone started at a high dose and then slowly tapered down has been reported to have beneficial effects on seizures and neurological functions in several series, particularly
when started early on in the course 27). For long term steroid therapy, it has been recommended to start with boluses of intravenous methylprednisolone [e.g. 400 mg/m2/day 28) or, in children, 20 mg/kg/day 29) and then to introduce 1–2 mg/kg/day oral prednisolone or prednisone 30). This dose should be slowly reduced, ideally to a dose below the threshold of Cushing’s syndrome.
Bahi-Buisson et al. 31) confirmed steroid treatment can be useful when given early on in the course of Rasmussen encephalitis, but they found that long term relapse can occur among the good responders requiring delayed hemispheric disconnection.
Immunoglobulins have also been used in Rasmussen’s as well immune mediated encephalitis 32). IVIg is a purified blood product pooled from many human donors composed mainly of IgG and some IgA. The precise mode of action of this product is unclear. Several studies have shown efficacy in treating patients with immunodeficiency. The use in patients with epilepsy has increased given the identification of immune mediated epilepsy but Cochrane reviews show no randomized evidence outside of specific syndromes such as anti NMDA and Landau Kleffner 33).
Steroid, immunoglobulins and other anti-inflammatory agent are also increasingly used in immune epilepsy. If these agents are to be used it would be important to Identify potential patients who have an immune basis for their seizures because adjunctive immunotherapy may slow, halt, or even reverse the epileptogenic process.
Zuliani 34) and Suleiman 35) have proposed guidelines for recognition of these patients in adults and children respectively. Clinical features suggestive of an autoimmune pathogenesis include patients with recent onset epilepsy (< 2 years), early antiepileptic drug resistance, and multifocal seizures and personal or family history of autoimmunity. Paraclinical findings suggestive of an autoimmune etiology include the detection of a neural autoantibody, inflammatory CSF (leukocytosis or CSF-exclusive oligoclonal immunoglobulin bands), or MRI characteristics suggesting inflammation (T2 hyperintensities, contrast enhancement on gadolinium studies, and/or restricted diffusion) and /or inflammatory neuropathology on biopsy.
Recurrent seizures are a common symptom in autoimmune neurologic disorders, especially in limbic encephalitis or multifocal paraneoplastic disorders. Autoantibody specificities reported in the setting of paraneoplastic limbic encephalitis include antineuronal nuclear antibody type 1 (ANNA-1), collapsin response-mediator protein 5 (CRMP-5), and Ma2. Autoantibodies with a commonly nonparaneoplastic etiology include Voltage-gated potassium channel (VGKC) complex or associated proteins including leucinerich glioma inactivated 1 (LGI1), contactin-associated protein 2 (CASPR2) and contactin 2 and glutamic acid decarboxylase 65 (GAD65) antibodies and have been reported in patients with limbic encephalitis and idiopathic epilepsy with antiepileptic drug-resistant seizures. More recently identified autoantibodies that strongly correlate with clinical seizures include N -methyl-D-aspartate receptor (NMDAR), 23 γ-aminobutyric acid B (GABAB) receptor, metabotropic glutamate receptor type 5 (mGluR5) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors.
No randomized clinical control trials on the use of corticosteroids in autoimmune epilepsies have been conducted to date. In an observational, retrospective case series by Quek et al. 36), neural autoantibodies were identified in 29/32 patients (91%). VGKC complex IgG antibodies were detected in 18/29 (62%) of which 14 were bound to LGI1 (78%), 1 was bound to Caspr2 and 3 were of unknown specificity. In addition, GAD65 was found in 7/29 (24%), and CRMP-5 was found in 2/29. In this study, 27 people underwent immunosuppressive treatment that comprised intravenous methylprednisolone alone (IVMP) (n = 12); intravenous immune globulin alone (IVIg) (n = 3); and combinations of IVMP, intravenous immune globulin alone (IVIg), cyclophosphamide, or plasmapheresis (n = 12). In 22/27 patients (81%), this therapeutic trial was positive with 18 patients becoming seizure free for at least 3 months and 4 patients having improved seizure frequency. Early treatment was associated with a favorable outcome.
Although strong evidence is lacking, the authors recommended that if autoimmune epilepsy is suspected, a trial of 6 to 12 weeks of immunotherapy (intravenous methylprednisolone or IVIg daily for 3 days and then weekly) is justifiable in the absence of other treatment options and may serve as additional evidence for an autoimmune etiology when a favorable seizure response is observed. They also recommended considering long-term immunosuppressive treatment, overlapping with gradual taper of intravenous methylprednisolone or IVIg, for patients whose seizures respond favorably to the initial trial of immunotherapy. Despite this, relapses may still occur.
Clinical trials
If other treatments don’t work or you are interested in exploring new therapies, taking part in an experimental clinical trial should be considered (https://www.epilepsy.com/clinical_trials). These trials may test a new medication, device, or surgical procedure that has not been approved by the FDA yet. Or there may be trials testing how well approved medications or therapies works compared to others.
Clinical research is also done to better understand the problems associated with uncontrolled epilepsy or how complementary therapies, such as diet, stress management, or safety devices may help.
While new therapies are developed by clinical trials, people need to be aware that they are participating in research. Supervision and safety controls are extensive, but there still is an element of risk and the unknown. If a trial is successful, you may get to use a new therapy years before it becomes available to the public.
Intractable epilepsy prognosis
Intractable epilepsy does not always remain intractable. First, one of the treatments listed above may prove effective. Second, individuals may be able to modify precipitating factors or their lifestyle to help to control the seizures. But even in the absence of specific therapies or life changes, there is hope for improvement. Jacqueline French and associates studied 246 patients from their clinic who had at least one seizure per month and were taking at least two seizure medications. Over a three-year follow-up period, 5% of these patients each year became seizure-free for at least six months. Unfavorable predictors of control were chronic cognitive delay, long history of intractable seizures and previous status epilepticus.
Despite the hope that some people will get better over time, you must also remember that uncontrolled seizures bring a number of other problems. People living with active seizures have greater risks of accidents, injuries, cognitive problems, mood disorders, social problems, unemployment and more. Unfortunately, serious and life-threatening risks of seizures are real. Understanding the causes and seriousness of uncontrolled epilepsy may help people get the right help as early as possible.
References [ + ]




{kind=link}