



CNS vasculitis
Central nervous system (CNS) vasculitis is the inflammation of blood vessels in the brain resulting in restricted blood flow. CNS vasculitis is a condition where the immune system attacks the blood vessels in the brain. CNS vasculitis is among a family of rare disorders characterized by inflammation of the blood vessels, which restricts blood flow and damages vital organs and tissues. A serious condition, CNS vasculitis can block the vessels that supply the brain and spinal cord, causing potentially life-threatening complications such as loss of brain function, or stroke.
CNS vasculitis can cause headaches, irritability, learning problems, vision problems, weakness on one side of the body, seizures, or stroke.
If you think your child may have CNS vasculitis, contact your family doctor and ask her to examine your child.
The naming of CNS vasculitis is very confusing. When you look at pages on the Web, you may see many different names for the same disease, including:
- Isolated angiitis of the CNS (IACNS)
- Transient cerebral arteriopathy (TCA)
- Transient cerebral vasculopathy
In medical journals, you will most often see childhood CNS vasculitis referred to as cPACNS. This stands for “childhood primary angiitis of the CNS.”
Different types of CNS vasculitis are described in these terms:
- the size of the blood vessel that is affected: small vessel or large vessel CNS vasculitis
- whether the CNS vasculitis is spreading (progressive) or not spreading (non-progressive)
CNS vasculitis is typically categorized as “primary” and “secondary”:
- Primary angiitis of the CNS (PACNS) is vasculitis confined specifically to the brain and spinal cord, which make up the central nervous system. It is not associated with any other systemic (affecting the whole body) disease.
- Secondary CNS vasculitis usually occurs in the presence of other autoimmune diseases such systemic lupus erythematosus, dermatomyositis, or rheumatoid arthritis; systemic forms of vasculitis, such as granulomatosis with polyangiitis, microscopic polyangiitis, or Behcet’s syndrome; or viral or bacterial infections.
Your doctor needs to find out which type your child has, because the different types are treated in different ways. In general, these are the three types:
- Small vessel CNS vasculitis: This type is severe and needs to be treated with strong medicine. Once it is found and treated, it heals very well.
- Progressive large vessel CNS vasculitis: This type will spread to other blood vessels if it is not treated.
- Non-progressive large vessel CNS vasculitis: This type does not spread, but it needs to be treated to prevent brain damage.
Diagnosis of CNS vasculitis can be challenging because a number of other diseases and infections have similar symptoms. Once diagnosed, CNS vasculitis is typically treated with corticosteroids such as prednisone, used in combination with medications that suppress the immune system. Even with effective treatment, relapse of CNS vasculitis is common, so ongoing medical follow-up is important.
Who gets CNS vasculitis?
In general, CNS vasculitis is considered rare. In the case of primary angiitis of the CNS (PACNS), the disorder can affect people of all ages but generally peaks around age 50. It most often occurs in males.
Primary CNS vasculitis of childhood
Primary CNS vasculitis of childhood is a serious but potentially reversible inflammatory brain disease. CNS vasculitis in children can occur as a primary disease that is isolated to the CNS or as a secondary manifestation of an underlying systemic condition. Although numerous systemic inflammatory diseases and infections have long been recognized as responsible for causing secondary CNS vasculitis, primary CNS vasculitis of childhood has only recently been described as a reversible inflammatory brain disease in case reports and case series 1).
To date no diagnostic criteria for children are available; thus, the adult definition is applied in practice 2). There are 2 main diagnostic categories: large-medium vessel disease and small vessel disease.
The diagnosis of large-medium disease is based on magnetic resonance angiography and conventional angiography evidence of vasculitis in the CNS, in the absence of underlying systemic inflammatory disease 3). Lesions typically conform to the territory defined by a particular arterial distribution.
Small vessel CNS vasculitis affects vessels smaller than those seen by magnetic resonance angiography and conventional angiography. Therefore, by definition, this condition has negative angiography findings 4). Lesions can be multifocal and bilateral and tend not to conform to a distinct vascular distribution. The diagnosis is confirmed by brain biopsy findings.
Large-medium vessel disease has been further subdivided into progressive and nonprogressive groups, which are defined by evidence of disease progression on angiography findings 3 months after diagnosis. Nonprogressive large-medium vessel vasculitis shares many similarities with both postvaricella angiopathy and transient cerebral arteriopathy of childhood. A significant overlap in the presentation and imaging findings among these conditions is often observed. Studies are needed to define the differences in pathophysiology and treatment requirements for each clinical disorder and to determine whether these are, in fact, distinct disease entities.
Children with progressive large-medium vessel disease typically have neurocognitive dysfunction at presentation, multifocal lesions on MRI, and evidence of distal stenosis on angiography 5).
The risk of progression in terms of new vascular lesions is considered to be low when the presentation consists of an isolated stroke and imaging reveals evidence of unilateral, proximal vessel stenosis 6). However, the risk of recurrent stroke due to the initial vascular injury in the same distribution is thought to be significant. Decreasing the subsequent stroke risk is the rationale for treating these patients with immunosuppressive therapy 7).
The cause of primary CNS vasculitis of childhood is unknown. By definition, any case of CNS vasculitis caused by an underlying systemic disease is not primary CNS vasculitis.
Primary Angiitis of the CNS
Primary angiitis of the central nervous system (PACNS) is frequently considered in the differential diagnosis in patients with cryptogenic neurologic illness or in young subjects with ischemic stroke. The absence of characteristic clinical, laboratory, or radiographic features of this rare disease make the diagnosis very difficult, and has contaminated the literature with unproven cases in which alternative diagnoses are plausible.
The causey and pathogenesis of primary angiitis of the CNS are unknown. The fundamental mechanism of all vasculitides is immunologic; Crowe 8) discussed 4 different mechanisms of tissue injury that might apply to the pathogenesis of vasculitis: immune complexes, direct antibody-mediated damage, delayed hypersensitivity, and cytotoxic T lymphocytes. With the limited knowledge scientists have about primary angiitis of the CNS, no strong evidence supports any of these mechanisms in the pathogenesis of this disease, although the granulomatous nature of inflammation suggests a role of cell-mediated immunity 9).
As in other autoimmune disorders, T cells that become sensitized in the course of systemic illness or viral infection probably later contribute to a cellular immune response directed against cross-reacting epitopes in CNS vessels 10). Other authors propose that, in the setting of altered host defense mechanisms, a virus or other pathogen may lead directly or indirectly to diffuse cerebral vasculitis 11).
The latter hypothesis is supported by the rare condition in which vasculitis involving mainly the ipsilateral anterior circulation with consequent infarcts occurs days to weeks following varicella-zoster infection of the first division of the trigeminal nerve. The mechanism seems to be retrograde spread of the virus from Gasserian ganglion to the arteries of the anterior circle of Willis 12).
Pathologically confirmed cases of primary angiitis of the CNS have been reported in patients with Hodgkin disease, amyloid angiopathy, and graft-versus-host disease. However, available information in these cases does not allow any conclusion about the causal relation between these diseases and primary angiitis of the CNS 13).
Regardless of the cause of primary angiitis of the CNS, the main mechanism of neurologic damage in these patients is ischemic. This results from 3 consequences of inflammation in the vascular wall: obstruction of the vessel lumen, increased local coagulation from the effects of proinflammatory cytokines on the endothelial surface, and alteration in vasomotor tone 14).
Primary angiitis of the CNS is reported more frequently in North America, Europe, Australia, and New Zealand. Whether the disease has a higher incidence in these regions or it has just been more successfully diagnosed and reported there is unclear.
Men are more commonly affected by primary angiitis of the CNS than women; the male-to-female ratio is about 7:3 15).
In most reported cases, patients were in the fourth to sixth decades of life at time of diagnosis. However, patients aged 7 months to 78 years have been described 16).
Mortality and morbidity of primary angiitis of the CNS are hard to determine due to the variability in diagnosis means and treatment among published series. However, treatment with steroids and immunosuppressants has improved the outcomes of the disease, which used to be fatal. In a recent report, 14% of 29 patients with biopsy-proven primary angiitis of the CNS died or had severe morbidity (Modified Rankin Scale of 5) at follow-up of 1.14 years 17).
CNS vasculitis causes
The brain and the spinal column make up the central nervous system (CNS). Vasculitis is an inflammation of the blood vessels. Blood vessels are the veins and arteries that carry blood around the body including the brain. So CNS vasculitis is an inflammation of blood vessels in the brain.
The cause of CNS vasculitis is not fully understood by researchers. Inflammation (irritation and swelling) is a normal process. It is how our immune systems protect our bodies from bacteria and viruses that cause infection. But in CNS vasculitis, there is no infection. Instead, the immune system wrongly attacks normal cells causing inflammation. This type of problem is called an autoimmune disease. CNS vasculitis is classified as an autoimmune disorder—a disease which occurs when the body’s natural defense system mistakenly attacks healthy tissues. Researchers believe an infection may contribute to the onset of CNS vasculitis. Environmental and genetic factors may play a role as well.
CNS vasculitis signs and symptoms
Many forms of vasculitis are accompanied by fever, fatigue, sudden weight loss, or skin rashes.
The first signs of CNS vasculitis are usually one or more of the following:
- Severe headaches that don’t go away 18)
- Irritability
- Learning problems
- Eyesight (vision) problems
- Problems dealing with loud sounds or bright lights
- Forgetfulness or confusion
- Abnormal sensations or loss of sensations
Most children are not diagnosed with CNS vasculitis when these symptoms first appear.
Usually, a child is diagnosed with CNS vasculitis after having one of these serious problems:
- Numbness, weakness, or paralysis of one side of the body (hemiplegia)
- Seizures or convulsions
- Strokes or transient ischemic attacks (mini-strokes)
- Swelling of the brain (encephalopathy)
- Difficulty with coordination.
The presentation may be affected by the type of vascular lesion (large-medium vs small vessel), as well as the location of discrete lesions seen on MRI and angiography.
- Large-medium vessel disease frequently presents with focal deficits, including acute hemiparesis, hemisensory deficit, or fine motor deficit. Diffuse deficits are also seen in this condition and may include headache, concentration and cognitive deficits, behavior and personality changes, and seizures. Neurocognitive dysfunction and headaches are reportedly more frequent in progressive large-medium vessel disease, whereas hemiparesis is a more common presentation in nonprogressive large-medium vessel disease 19).
- Small vessel disease also has a wide variety of presentations, including seizures (either acute onset or a chronic seizure disorder), headache, neurocognitive deficits, and psychiatric symptoms, including psychosis. Focal deficits that involve gross motor skills, fine motor skills, and sensory function are also seen 20).
- Constitutional symptoms are uncommon in patients with large-medium vessel disease but can be present in a minority of patients with small vessel disease. These may include fever, fatigue, and flulike symptoms. In general, such systemic symptoms should prompt investigation for infection or other secondary cause of CNS vasculitis.
- Eliciting features of systemic inflammatory disease that exclude the diagnosis of primary CNS vasculitis is important. These may include rashes, arthritis, respiratory symptoms, urinary abnormalities, and gastrointestinal symptoms among others.
CNS vasculitis complications
CNS vasculitis can harm the brain.
The inflammation caused by CNS vasculitis can harm the brain. When they are inflamed, the walls of the blood vessels get thicker and the space inside them gets smaller. This means that less blood can flow through them. Parts of the brain get less blood than they need. Sometimes they get no blood at all. When this happens, two major problems can result:
- Brain tissue around the inflamed blood vessel becomes irritated or damaged. This is also called brain inflammation.
- Brain tissue around inflamed blood vessels does not get enough oxygen. This can cause a stroke.
Complications related to disease, such as ongoing seizure disorder, may be noted. Flare of the disease is possible even while receiving therapy. The neurological signs that accompany this may be subtle, and repeating neuroimaging to ascertain the presence of new lesions on MRI or conventional angiography is often necessary. This may require increasing current immunosuppression or the institution of a different immunosuppressive medication to induce remission.
CNS vasculitis diagnosis
Diagnosing CNS vasculitis poses a challenge for physicians. Many of the key symptoms of CNS vasculitis are shared by other diseases and infections, so these “mimics” must be ruled out. There is no single diagnostic test for CNS vasculitis, so your doctor will consider a number of factors, including a detailed medical history, a physical examination, laboratory tests, and specialized imaging studies. A biopsy of tissue from blood vessels in the brain or spine is usually required to confirm a diagnosis.
If CNS vasculitis is suspected, your doctor will likely order the following tests:
- Lab work: Blood tests are frequently normal in PACNS vasculitis, but may be abnormal if reflecting another underlying disease.
- Examination of the spinal fluid: A sample of the cerebrospinal fluid (which surrounds that brain) is removed through a spinal tap and analyzed for infection and signs of inflammation.
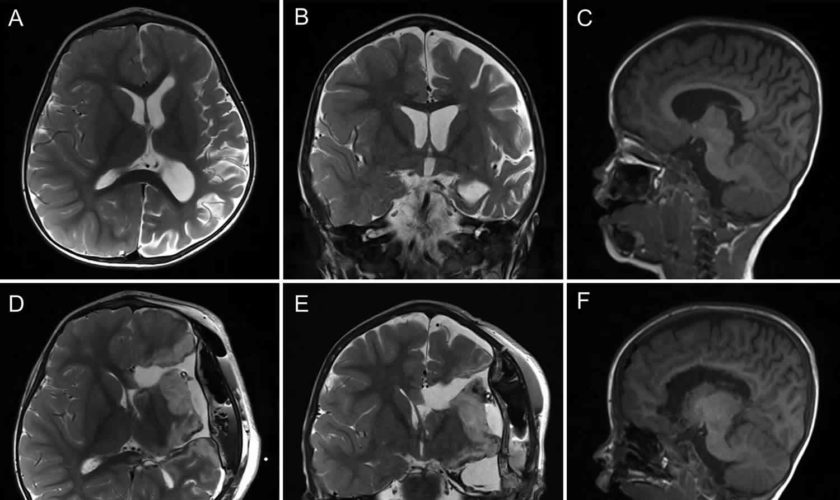
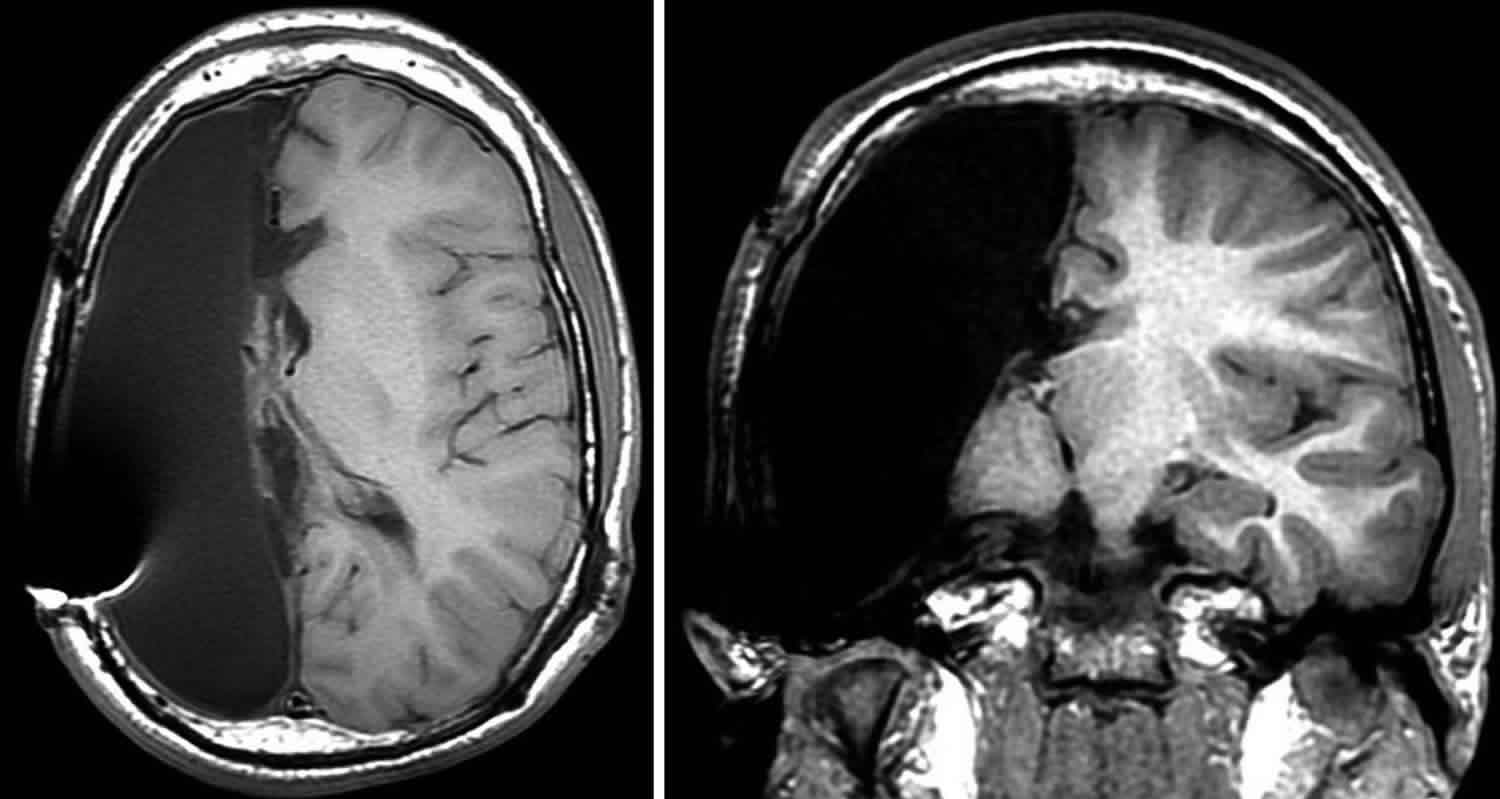
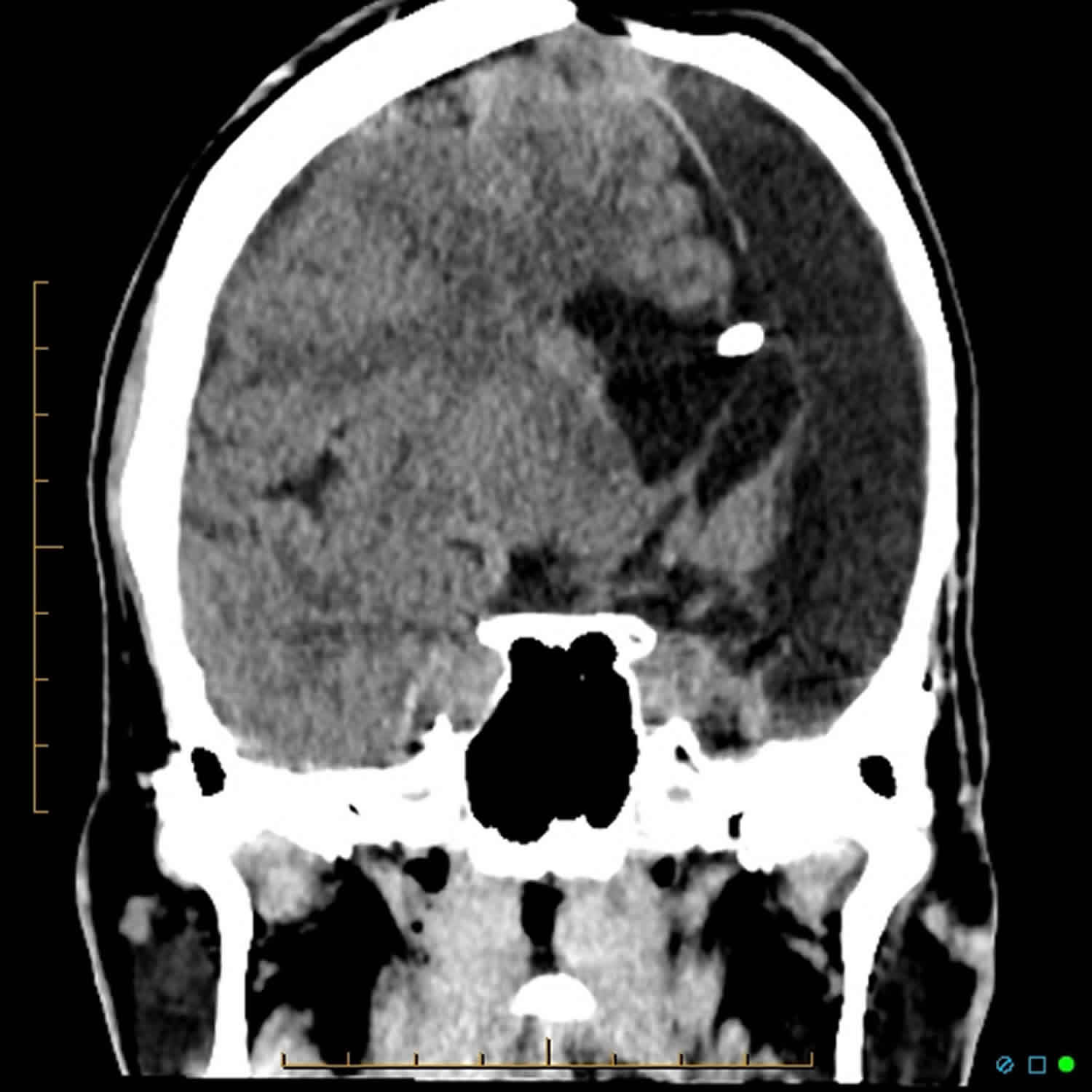
- Diagnostic imaging: Computed tomography (CT) scans and magnetic resonance imaging (MRI) produce images that can help identify abnormalities of the brain, spinal cord, blood vessels, and other organs and tissues.
- Cerebral angiogram: An angiogram detects blockages of blood vessels using X-rays taken during the injection of a contrast agent.
- Biopsy: This surgical procedure removes a small tissue sample from a blood vessel or an affected organ, which is examined under a microscope for signs of inflammation or tissue damage. Because other conditions can cause similar brain vessel abnormalities as CNS vasculitis, a brain biopsy may be the only way to make a definitive diagnosis.
Medical history and physical exam
First, the doctor will ask about your child’s symptoms and medical history. The doctor will do a physical exam to see if your child has any symptoms of brain inflammation.
Blood sample and spinal tap
Next, the doctor will take samples of your child’s blood and cerebrospinal fluid (CSF). CSF is the fluid that surrounds the brain and spinal cord. To get a sample of CSF, your child will need to have a spinal tap (lumbar puncture).
Your child’s blood and CSF will be tested to see if they contain certain substances that can show if your child has inflammation somewhere in the body.
MRI scan
Next, the doctor will order an MRI scan. This test makes special pictures of the brain, using radio waves and a strong magnet. The doctor will look at these pictures to see if your child’s brain has either of these:
- areas of brain inflammation
- areas where not enough blood is flowing (ischemia or stroke)
Angiograms
If the MRI shows problems, the doctor will order two kinds of angiogram. An angiogram is a test that gives pictures of the large blood vessels. The two types are as follows:
- A magnetic resonance angiogram (MRA) uses the same machine as an MRI. It gives a 3-dimensional picture of the large blood vessels.
- An X-ray angiogram gives a picture of the large blood vessels. Your child will have a special dye injected into his blood. This dye is called contrast fluid. It helps blood vessels show up on the X-ray.
Angiograms will show if your child has large vessel CNS vasculitis. But they do not show the small blood vessels. So even if these tests do not show any problems, your child could still have small vessel CNS vasculitis.
Brain biopsy
The only way to find small vessel CNS vasculitis is with a test called a brain biopsy. A neurosurgeon will take a tiny sample from your child’s brain. The sample is only the size of a needle. Another doctor will look at it under a microscope.
This test will show if your child has small vessel vasculitis. It can also help tell CNS vasculitis apart from other diseases, such as infections or brain tumors.
A major mimic of CNS vasculitis
Reversible cerebral vasoconstriction syndrome refers to a group of conditions that involve spasm of the brain vessels, and that mimic CNS vasculitis. Reversible cerebral vasoconstriction syndrome features sudden, severe headaches, as well as strokes or bleeding into the brain. It is essential for clinicians evaluating patients to be aware of reversible cerebral vasoconstriction syndrome and to distinguish it from CNS vasculitis, given the differences between the two diseases, including the treatment and prognosis.
CNS vasculitis treatment
CNS vasculitis is typically treated with a high-dose corticosteroid, such as prednisone, to reduce inflammation. For more severe cases, prednisone is used in combination with drugs that suppress the immune system’s response, such as cyclophosphamide, mycophenolate mofetil or azathioprine. Treatment may be aggressive for the first six months and then tapered down as symptoms improve. A full course of treatment takes about two years.
The treatment of CNS vasculitis aims to do these things:
- improve the blood supply to the brain
- prevent further complications
- prevent blood clots from forming
CNS vasculitis is treated with medicine that stops the immune system from working so hard. This stops the inflammation and protects the brain. Your child will also need to take medicine to prevent blood clots.
Your child will need to take different kinds of medicine for CNS vasculitis:
- Medicine to suppress the immune system and reduce the inflammation in the blood vessel walls. This includes prednisone, cyclophosphamide, azathioprine and mycophenolate mofetil.
- Medicine to make the blood thinner and prevent blood clots. This includes low-dose ASA (acetylsalicylic acid) and heparin.
Your child will be taking these medicines for many months. You need to know about them and about the side effects that your child may have. These are described in more detail below.
If you are worried about any side effects, talk to your child’s doctor.
In addition to medication, other forms of treatment may include physical, occupational or speech therapy. If memory is affected, brain activities that enhance memory may be recommended.
Heparin
Children with large vessel CNS vasculitis often take intravenous (IV) heparin at first. Intravenous means your child will take this medicine through a needle or a tube that puts it directly into their bloodstream.
Later, your child may take low molecular weight heparin (LMWH). This medicine is given with a needle under your child’s skin.
Acetylsalicylic acid (ASA)
If your child has large vessel CNS vasculitis, they will also take ASA (acetylsalicylic acid) for the whole time they are being treated. ASA makes your child’s blood thinner. This helps your child’s blood travel through the blood vessels.
Your child will take a very low dose of ASA, between 2 and 5 mg per kilogram of body weight per day. At this dose, ASA has no major side effects. Some children may have an upset stomach.
Prednisone
Prednisone is a drug that suppresses the immune system. This means that it lowers the number of immune cells that are attacking your child’s blood vessels. When your child takes prednisone for a long time, the immune system is able to “reset” itself and make a new set of healthy immune cells. The new immune cells will not attack your child’s body.
Your child will be taking a high dose of prednisone to start with. As the treatment goes on, the doctor will lower the dosage.
Your child needs to take the amount of prednisone that the doctor prescribes. This is very important. If your child is taking high doses of prednisone and then suddenly stops, your child’s body will not be able to adjust to the change.
The side effects of prednisone are not pleasant, but there is no other way to treat CNS vasculitis. The effects are stronger at higher doses and with longer courses of treatment. The main side effects are:
- weight gain
- more risk of getting infections
- hair growth
- stretch marks
- acne
- mood swings
- upset stomach
- higher blood pressure
- higher blood sugar
- thinning bones (osteoporosis)
These side effects are all temporary. They will go away as soon as your child stops taking the prednisone.
Cyclophosphamide
Cyclophosphamide also suppresses the immune system. It slows down or stops the growth of immune system cells. Your child will take cyclophosphamide once a month through an IV tube.
Cyclophosphamide is also used to treat some kinds of cancer. But we use much lower doses for CNS vasculitis. This means that CNS vasculitis patients have fewer side effects than cancer patients.
For children with CNS vasculitis, cyclophosphamide has three main side effects:
- more risk of getting infections
- more risk of bleeding
- upset stomach and vomiting (throwing up)
A very large dose of cyclophosphamide can make it hard for girls or boys to have children later on. We will watch your child’s treatment to make sure they do not reach this dose.
Azathioprine (Imuran)
Azathioprine also suppresses the immune system. It stops immune system cells from dividing.
Common side effects include:
- stomach upset
- diarrhea
- allergic reactions
- a flu-like illness
- liver irritation
To watch for problems, your child will need a blood test every month.
Some people cannot remove azathioprine from their bodies. Before your child starts taking this medicine, they will need a blood test to find out if they can take it safely.
Mycophenolate mofetil (Cellcept)
Mycophenolate mofetil (MMF) also suppresses the immune system. It stops immune system cells from getting overactive.
The most common side effect is stomach upset. To prevent this, your child should take the medicine with food.
A more serious side effect is when mycophenolate mofetil stops the body from making white blood cells, red blood cells and platelets. To watch the levels of these blood cells, your child will need regular blood tests.
Medication side effects
The medications used to treat CNS vasculitis have potentially serious side effects, such as lowering your body’s ability to fight infection, and potential bone loss (osteoporosis). Side effects of prednisone, such as weight gain, susceptibility to infection, hypertension, and osteopenia, can be seen with the prolonged course of corticosteroids that are a mainstay of treatment. Therefore, it’s important to see your doctor for regular checkups. Medications may be prescribed to offset side effects. Infection prevention is also important. Talk to your doctor about getting a flu shot, pneumonia vaccination, and/or shingles vaccination, which can reduce your risk of infection.
Preventing infection during treatment for CNS vasculitis
The medicines for CNS vasculitis make the immune system less active. This means your child is more likely to get sick if they come in contact with germs. To help keep your child and family healthy:
- Wash hands often.
- Stay away from anyone with an illness that is catching.
- Keep all family members’ immunizations up to date.
Remember, your child’s immune system is still working. Your child does not need to “live in a bubble.” Your child can still have a normal lifestyle and stay healthy.
Do not be afraid to ask your child’s doctor if you have any questions.
Immunizations
While your child is being treated for CNS vasculitis, they should not take live vaccines. One common live vaccine is the MMR (measles, mumps and rubella) vaccine.
Ask your doctor which immunizations are safe for your child to have.
Diet
- A healthy diet with low fat and sodium intake is appropriate when a patient begins corticosteroid treatment.
- Adequate intake of calcium and vitamin D, with supplementation when necessary, is essential when children are treated with corticosteroids.
Further outpatient care
Ongoing close follow-up with a multidisciplinary team is important in primary CNS vasculitis of childhood. The particular needs of each patient should be identified and addressed as they arise during treatment. These may include educational support for reintegration in school, adequate seizure control, and emotional support of the family, among others.
- In both small vessel disease and large-medium vessel disease, MRI should be repeated at 3 months and 6 months following disease onset to study the changes in parenchymal brain lesions. In large-medium vessel disease, little improvement in the vessel anatomy may be observed until 6 months after diagnosis, at which time conventional angiography should be performed. Of course, should clinical symptoms change or disease progression be suspected, early imaging is appropriate.
- Once a diagnosis of small vessel disease is confirmed by biopsy findings, further biopsies do not need to be performed.
- The involvement of psychiatrists to assist with behavioral symptoms secondary to inflammatory brain disease is often necessary; psychiatric medication may be needed.
- Serial cognitive assessments with the Pediatric Stroke Outcome Measure are a useful way to quantify deficits.
- Annual neuropsychological assessments for evaluation of cognitive deficits and identification of assistance needed in school should be performed.
- Structured quality-of-life assessments using standardized questionnaires can provide insight into the impact of disease on a child’s daily life.
Your child will be monitored during treatment
MRIs and angiograms
Your child will have MRI tests often during treatment. Your child may also have angiograms. These tests help doctors see how the blood vessels are healing. This lets them make sure your child is getting the right amount of medicine.
Blood tests
Your child will have regular blood tests to see how the medicine affects her immune system. The goal is to keep your child’s immune system low enough to stop it attacking the blood vessels, but not so low that your child is at risk for serious infections.
Tests of thinking and learning
When your child is diagnosed with CNS vasculitis, a psychologist will test her thinking and learning skills. Your child will be tested again every year to see if there are any changes. If necessary, the psychologist will work out a plan so that your child’s school can help her learn better.
Your observations
Probably the most important way to monitor CNS vasculitis is through what you notice about your child. Your child will come for regular clinic visits. At these visits, the doctor will ask you and your child about any changes in behaviour that you have noticed at home or at school.
Here are some examples of things to watch:
- mood
- attention span
- ability to perform daily tasks at home and at school
Remember, you know your child best. If anything seems unusual or important, mention it to your doctor.
CNS vasculitis relapse
Even with effective treatment, relapses are common for individuals with CNS vasculitis. If your initial symptoms return or you develop new ones, report them to your doctor as soon as possible. Regular check-ups and ongoing monitoring of lab and imaging tests are important in detecting relapses early.
Living with CNS vasculitis
Living with a chronic disease such as CNS vasculitis can be overwhelming at times. Fatigue, pain, emotional stress and medication side effects can take a toll on your sense of well-being, affecting relationships, work and other aspects of your daily life. Sharing your experience with family and friends, connecting with others through a support group, or talking with a mental health professional can help.
CNS vasculitis prognosis
There is no cure for CNS vasculitis at this time, however it is treatable. Early diagnosis and treatment are essential to prevent potentially life-threatening loss of brain function or stroke. Other diseases often have the same symptoms as CNS vasculitis, so accurate diagnosis involves ruling out these conditions. Even with treatment, relapses are common with CNS vasculitis, so follow-up medical care is essential.
With early recognition and prompt treatment, prognosis can be excellent 21). Neurological recovery may take place over months, during which time physical therapy, occupational therapy, speech language therapy, appropriate schooling, and seizure control should be continued. Families of patients affected with primary CNS vasculitis should be prepared for a prolonged rehabilitation period.
Scientists do know that children’s brains have more ability than adults’ brains to “rewire” themselves. This means they can sometimes find new pathways to replace a damaged area. Although many patients exhibit complete neurological recovery, deficits may remain after completion of treatment. This may include focal and diffuse neurological deficits and behavioral and cognitive symptoms.
References [ + ]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}