




Toe walking
Toe walking is a pattern of walking in which a child walks on balls of his or her feet, with no contact between the heels and ground. Most children begin walking at 12 to 15 months of age. Toe walking is common in children who are learning to walk. When children start to learn walking, they try different foot positions, and walking on their toes may be part of this. After the age of 2, however, most children outgrow toe walking and begin to walk with their feet flat on the ground (a normal heel-to-toe pattern). By 3 years of age, children should walk with a heel-toe pattern.
Most children begin walking at 12 to 14 months with their feet flat on the ground. However, there are some children who begin walking on their tip toes instead. This pattern normally disappears within three to six months of learning how to walk. It almost always is completely gone by the end of the third year.
In very rare cases, continuing to toe walk after age 2 may be a sign of an underlying medical condition. In the vast majority of cases, however, persistent toe walking is “idiopathic” which means that the exact cause is not known. A non-idiopathic cause may be cerebral palsy, autism, sensory processing disorder, muscular dystrophy or brain injury. As children learn to walk, some toe walking is to be expected. When this becomes a strong habit that they do not grow out of, or the predominant pattern as they are new walkers, then several issues can arise.
Older children who continue to toe walk may do so simply out of habit or because the muscles and tendons in their calves have become tighter over time.
The following are negative consequences of toe walking:
- Tight ankles or contractures can develop
- Poor balance reactions, frequent falling
- Muscle imbalances “up the chain” meaning decreased hip or core strength due to the different postural alignment
- Difficulty with body mechanics including squatting or performing stairs, secondary to tight calve muscles
- Inability to stand with heels flat on the ground
- Pain in ankles, knees or hips due to faulty mechanics
- Surgery, casting, night splinting or daily bracing may be necessary
While some toe walking should not be alarming, the earlier you intervene, the better. Discuss this with your pediatrician or see a physical therapist who can provide early strategies to stop the cascade of effects that can be seen later.
Idiopathic toe walking in children is not a serious condition. It often resolves spontaneously and does not cause the child significant problems apart from the cosmetic appearance. Normally, your child will not need surgery. In addition to stretching and strengthening, treatments may include repeated casting of feet and ankles, bracing devices, or a combination of the two. More recently, the injection of Botulinum Toxin A (Botox) has been used to weaken the calf muscles, thus preventing tip toe walking. You can discuss these treatment options with your physician. It is important to understand that even though your child may achieve short-term improvement in muscle length and ankle range of motion, these treatments may not always guarantee a normal heel-to-toe walking pattern.
Toe walking key points
- Idiopathic toe walking is when a child continues to walk on their tip toes beyond three years of age.
- Idiopathic toe walking can lead to tight calf muscles and decreased movement of the ankles.
- Treatment for children younger than six years of age include calf stretches, Achilles tendon stretches and sit to stand exercises.
- Treatment for children six years of age and older include calf stretches and other exercises including marching on the spot, walking uphill and on uneven surfaces, heel walking and squats.
See your doctor if your child is toe walking AND:
- Walks on her toes most of the time
- Has stiff muscles
- Is uncoordinated
- Walks awkwardly and stumbles all the time
- Has fine motor skills that don’t seem to be developing normally (for example, she can’t button her shirt)
- Seems as though she can’t bear her weight on a flat foot
- Loses motor skills she already had
- Has any other medical problems
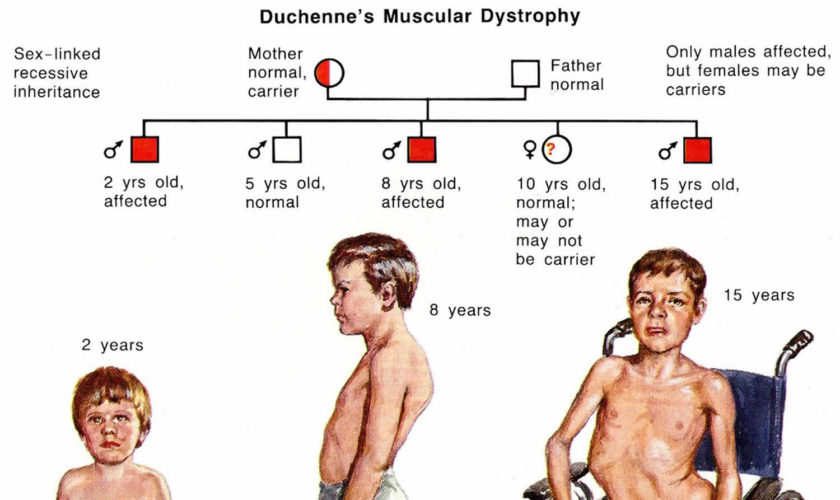
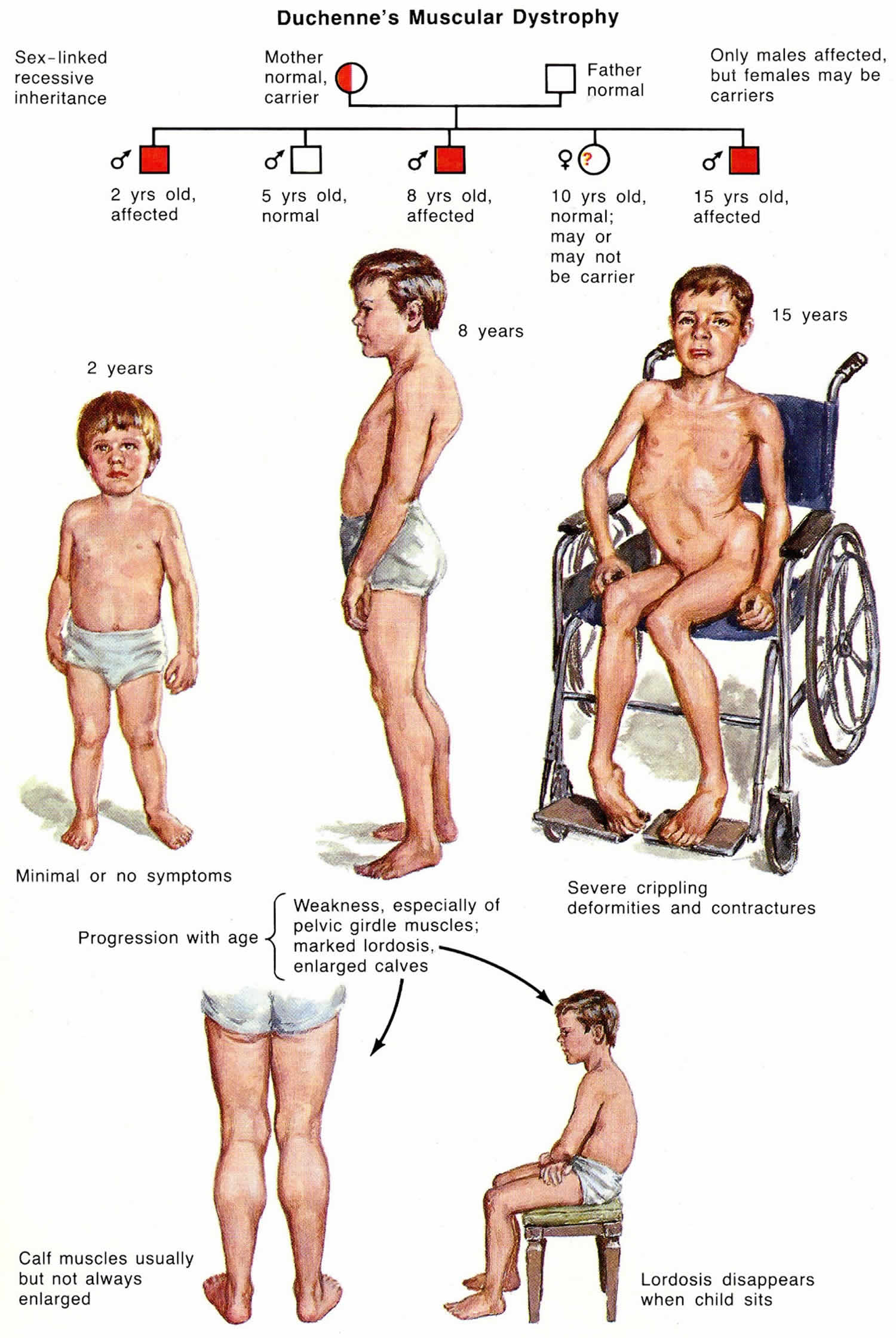
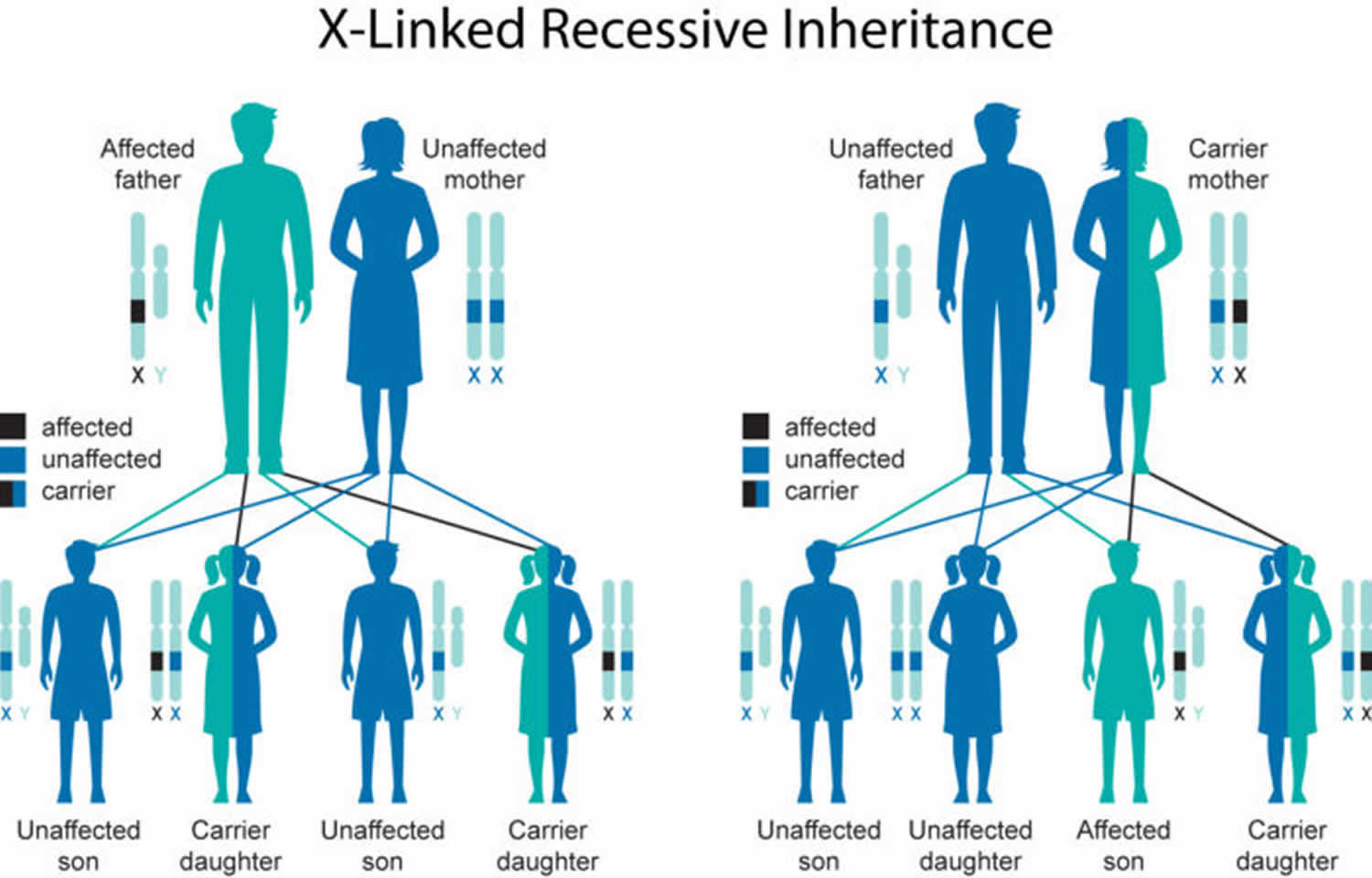
- Has a family history of muscular dystrophy or autism
- Was born prematurely
- Has previously walked flat-footed and only recently began to toe walk
- Avoids eye contact or exhibits repetitive behaviors such as rocking or spinning
Is toe walking just a developmental variation?
Generally, until age 2, toe walking isn’t something to be concerned about. Often, children who toe walk after that do so out of habit. More than half of young children who toe walk will stop doing so on their own by about age 5. Most children toe walk occasionally when they’re cruising around a room (by holding on to furniture), especially if they’re on a bare floor. Some kids keep toe walking, off and on, just for fun. Toe walking out of habit, also known as idiopathic toe walking, sometimes runs in families. The cause of idiopathic toe walking is unknown.
Parents of more than 1,400 children participated in a study conducted in Blekinge County in southeast Sweden. The results, published in Pediatrics in 2012, showed more than half of young children who toe walk stopped doing so on their own by about age 5, and most toe walkers did not have any developmental or neuropsychiatric problems.
Toe walking anatomy
The calf is formed by two major muscles. They are:
- Gastrocnemius muscle. This is the larger calf muscle. Its two parts form the bulge that is visible beneath the skin.
- Soleus muscle. This smaller, flat muscle lies underneath the gastrocnemius muscle.
Both muscles merge at the base of the calf, where they transition into becoming the Achilles tendon. The Achilles tendon then inserts into the calcaneus (heel bone). When you contract your calf muscles, the Achilles tendon pulls on your heel.
In some children who toe walk, this muscle-tendon combination may be shorter at birth, or may shorten over time, which prevents the child from touching his or her heels to the ground and walking flat-footed. However, in most children who toe walk, the muscle-tendon combination is long enough that the child is able to walk with his or her heels down if reminded to do so.
Figure 1. Toe walking anatomy
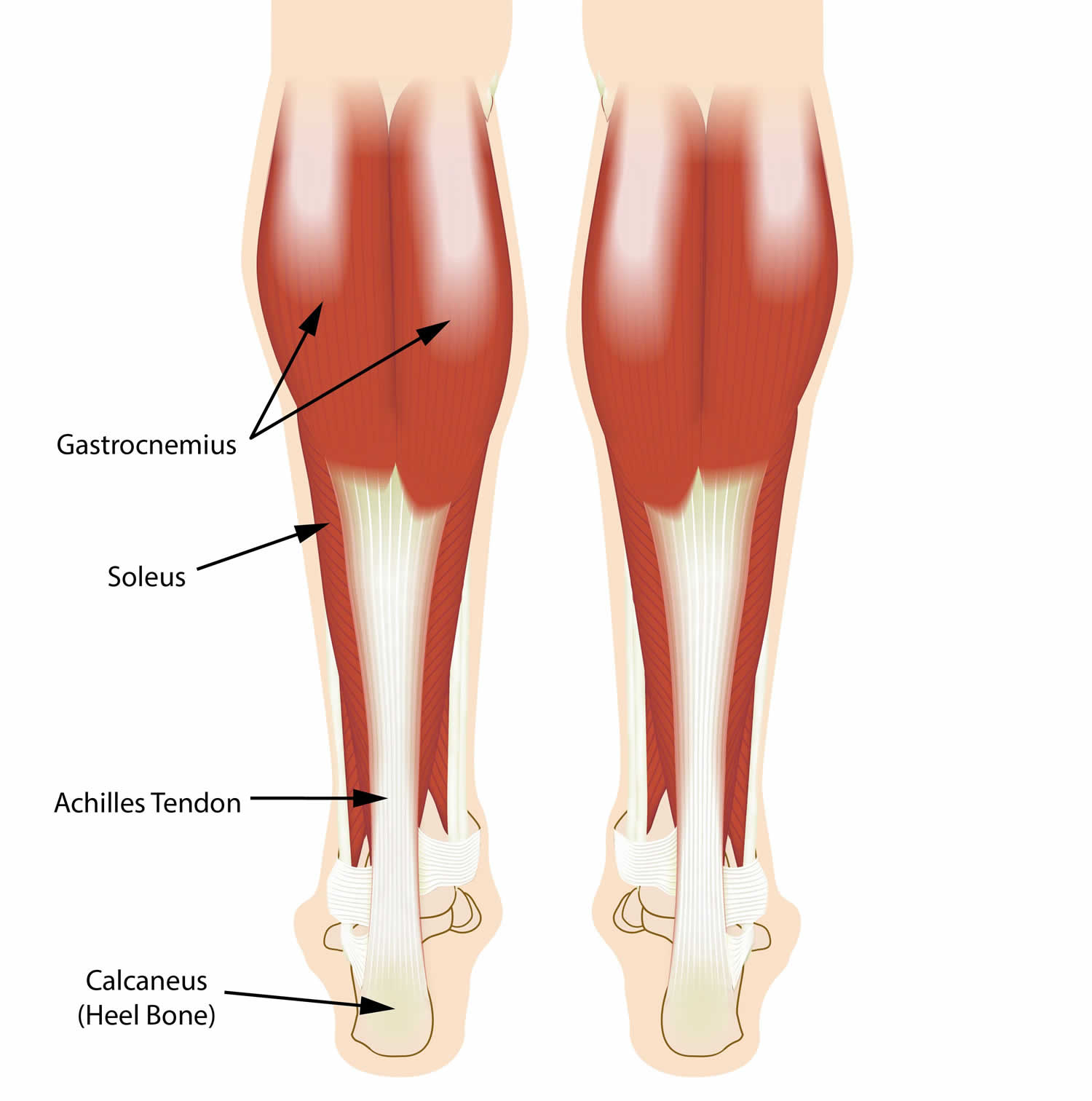
Footnote: The calf muscles and Achilles tendons work together to help lift your heels when you walk.
Toe walking causes
In the vast majority of children, toe walking is “idiopathic,” which means that the exact cause is unknown. When these children are evaluated by a doctor, their physical exams and neurological tests are normal.
Rarely, toe walking can be the result of a short Achilles tendon (the tendon that links the lower leg muscles to the back of the heel bone), cerebral palsy, muscular dystrophy, or another generalized disease of nerve and muscle. Toe-walking can also be associated with sensory-processing disorder. Sensory processing happens constantly every time we eat, move, or do an activity. Problems arise when the brain is unable to organize this information appropriately – like a traffic jam of information. Sensory-processing disorder can manifest itself in a myriad of ways, depending on which senses are affected. Children with autism also may walk on their toes or the balls of their feet. But as long as your child is growing and developing normally, toe walking in its own is unlikely to be a cause for concern.
Idiopathic toe walking
Toe walking out of habit, also known as idiopathic toe walking, sometimes runs in families. Idiopathic toe walking is when a child continues to walk on their tip toes beyond three years of age. They will often stand with their feet flat on the ground, but when walking or running will prefer to be on their toes. If your child does not outgrow tip toe walking by three years of age, take them to see a health-care professional.
Medical causes
In a smaller number of cases, persistent toe walking can be a sign of an underlying medical condition, such as:
- Cerebral palsy. Toe walking can be caused by a disorder of movement, muscle tone or posture caused by injury or abnormal development in the parts of the immature brain that control muscle function.
- Muscular dystrophy. Toe walking sometimes occurs in this genetic disease in which muscle fibers are unusually prone to damage and weaken over time. This diagnosis might be more likely if your child initially walked normally before starting to toe walk.
- A spinal cord abnormality
- A short Achilles tendon. This tendon links the lower leg muscles to the back of the heel bone. If it’s too short, it can prevent the heel from touching the ground.
Although children with autism-related conditions toe walk more frequently than children who are developing normally, there is no direct link between the two conditions, and their toe walking may be sensory-related.
Toe walking signs and symptoms
Although doctors do not really know why some children prefer to walk on their toes, they do know that idiopathic toe walkers:
- walk on tip toes on both sides
- are constantly balancing on their toes
- are physically able to keep up with other children their age
- walk with straight knees
- will often be able to stand with their feet flat on the ground
- often have a family history of toe walking
Most young children who walk on their toes are able to walk flat-footed when asked to do so. However, many older children who continue to toe walk (usually those over the age of 5 are not able to walk with their heels down. These children may complain about problems wearing shoes or participating in sports or recreational activities that involve wearing roller skates or ice skates.
Some children who toe walk have no specific complaints, but their parents are still concerned about the impact their walking pattern may have on their future function as teenagers and adults.
Toe walking complications
Children who walk on their toes can develop tight calf muscles on the backs of their legs and have decreased movement of their ankles. In addition, the muscles on the front of their legs may become weak. If there is tightness and weakness, your child will have difficulty walking on their heels. Early identification of toe walking can help lead to the prevention of these muscle problems.
Kids who develop stiffness, tightening, and pain in their Achilles tendon can be treated with physical therapy and stretching exercises. Rarely surgery may be required (usually after age 6) if the toe walking is the result of (or results in) tendon stiffness.
Toe walking diagnosis
Your child’s doctor will begin by asking a number of questions, including:
- Were there any pregnancy complications or was your child born prematurely?
- How old was your child when he or she reached developmental milestones such as smiling, sitting, and walking?
- When did the toe walking start? (For example, did it begin when your child started to walk independently or at an older age?)
- Is the toe walking on both sides or only on one side? (Toe walking on just one side may be more concerning to your doctor since it can sometimes indicate a neurological problem.)
- Is there a family history of toe walking?
- What percentage of time is spent walking on the toes?
- If asked, is your child able to walk flat-footed?
- Does your child complain of foot or leg pain, weakness in the legs, or difficulty keeping up with children the same age?
Physical examination
The physical exam will typically begin with your doctor observing your child walk. In order to avoid the “doctor walk” (the patient does his or best to walk properly when the doctor is watching), this may be done even before your child realizes that he or she is being watched.
Your doctor will then ask to see your child’s typical walk (on the toes), followed by his or her “best” walk (walking as flat-footed as possible). In addition to observing the toe walking itself during this time, your doctor will also be evaluating the smoothness of the walk as part of a neurological evaluation.
During the physical exam, your child’s doctor will also:
- Check your child’s feet for abnormalities, including differences between the left foot and right foot.
- Look for differences in leg length and in the size of the thighs and calves.
- Assess if one or both calf muscles are tight by asking your child to move his or her feet and ankles in a number of different ways.
- Check range of motion in the hips and knees.
- Look for any skin abnormalities in the lower extremities and lower back.
Tests
Neurological exam. Some simple neurological tests will help determine if abnormalities in your child’s nervous system could be contributing to the toe walking. The exam will be tailored to your child’s age, developmental level, and ability to cooperate.
During the exam, your child’s doctor will:
- Assess if there is any contracture or excessive tightness of the muscles in the arms or legs.
- Check the strength of the major muscles.
- Check your child’s reflexes by tapping a small rubber hammer or a fingertip on different points on the body.
- Test sensation, or feeling, in the arms and legs.
Other tests. Idiopathic toe walking is a diagnosis of exclusion, meaning that no other problems can be identified from your child’s medical history and physical exam. For this reason, specific tests—such as x-rays, CT and MRI scans, and nerve and muscle tests involving electrode patches or needles electromyography (EMG)—are not usually ordered.
During an EMG, a thin needle with an electrode is inserted into a muscle in the leg. The electrode measures the electrical activity in the affected nerve or muscle.
If the doctor suspects a condition such as cerebral palsy or autism, he or she may recommend a neurological exam or testing for developmental delays.
Toe walking treatment
Treatment for toe walking depends on a number of factors, including:
- The age of your child
- Whether your child is able to walk flat-footed
Treatment for persistent toe walking often involves a period of casting or bracing to help stretch the muscles and tendons in the calves and encourage a normal gait.
Nonsurgical treatment
For children who are 2 to 5 years old and able to walk flat-footed, initial treatment is always nonsurgical.
Nonsurgical treatment may include:
- Observation. Your doctor may recommend simply monitoring your child with regular office visits for a period of time. If he or she is toe walking out of habit, it may stop on its own.
- Serial casting. Your doctor may apply a series of short leg walking casts to help progressively stretch and lengthen the muscles and tendons in the calf and break the toe-walking habit. Serial casting is usually performed over a period of several weeks.
- Bracing. Wearing an ankle-foot orthosis (AFO) can help stretch and lengthen muscles and tendons. An AFO is a plastic brace that extends up the back of the lower leg and holds the foot at a 90 degree angle. Typically, bracing is performed for a longer period of time than casting (months rather than weeks).
- Botox therapy. For certain patients—usually those with a neurologic abnormality that leads to increased muscle tone—an injection of botulinum A toxin (Botox®) may also be given to temporarily weaken the calf muscles. This will allow the muscles to stretch more easily during casting or bracing.
Shoes for your child
Wearing shoes may not correct toe walking. However, appropriate foot wear can help your child bring their heels further down. When selecting shoes for your child, keep in mind the following criteria:
- Choose a high cut shoe with a wide sole which provides good foot support.
- The shoe should be rigid or firm, not flexible in the middle section.
- The back of the heel should be firm.
Home exercises
If your child has idiopathic toe walking, a daily home exercise program can be very helpful. The goal is to stretch the calf muscles and strengthen the muscles on the front of the legs. This will help your child to be able to successfully walk with a heel-to-toe pattern.
If your child’s calf muscles are tight, or ankle motion is limited, you will be shown stretches to do at home with them. These stretches should be followed with activities to help them use their muscles in their new lengthened position.
These exercises will be necessary and beneficial as long as your child demonstrates a tip toe walking pattern. The exercises will vary with their age. The most important part of the exercise program is to remember to have fun with your child!
Stretches and strengthening exercises for children under six years of age
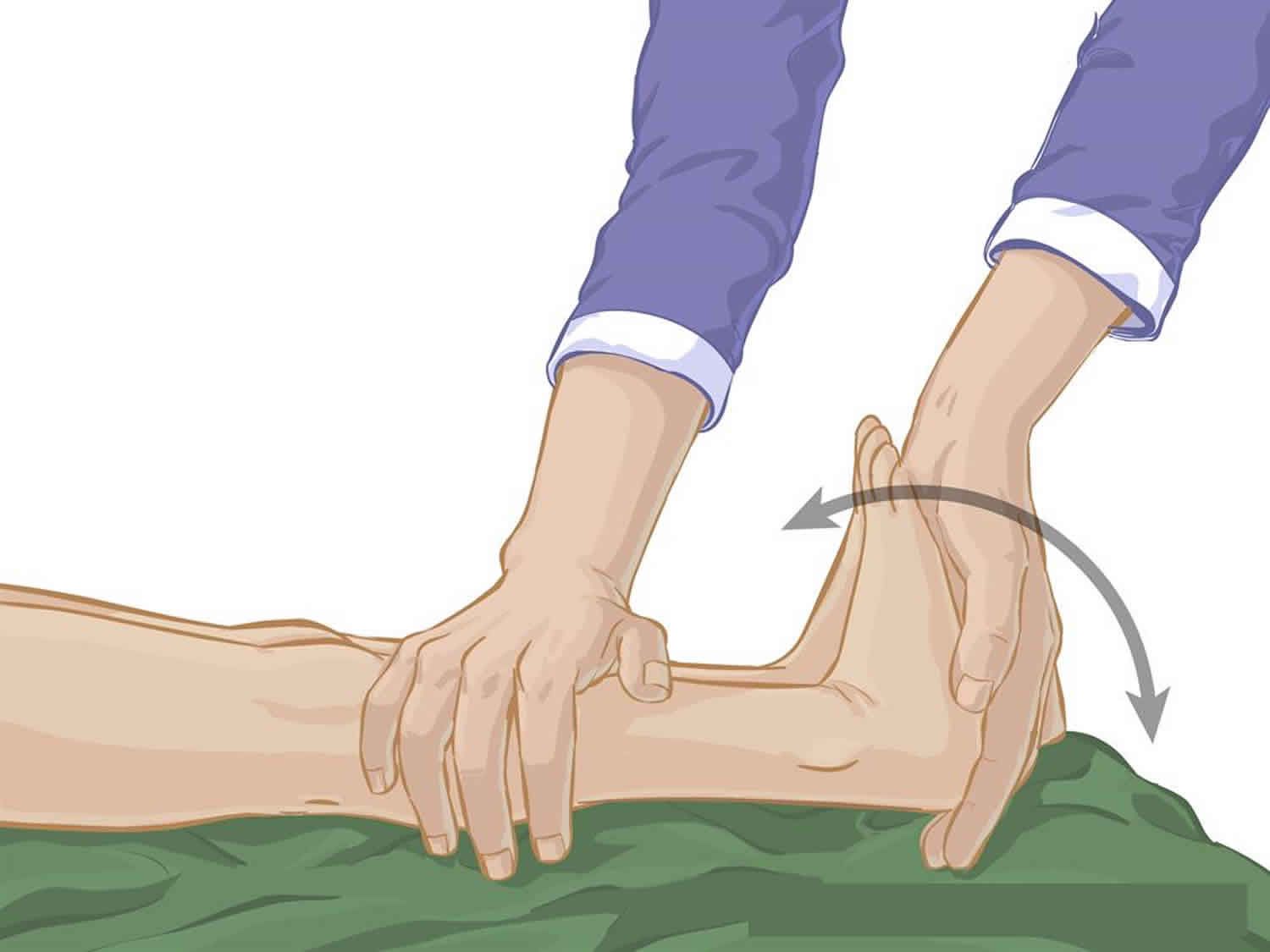
Calf stretch
- Have your child lie on their back on a comfortable surface such as a firm bed.
- With their knee straight and leg supported on the bed, bring your child’s foot upwards, toward their head, bending their ankle.
- Hold the stretch at the end of the movement (that is, as far as your child’s range of motion will permit) for 15 to 30 seconds. This should not be painful for your child.
- Bring your child’s foot back to a normal position. Repeat the exercise 10 times on each leg, daily.
Figure 2. Calf stretch
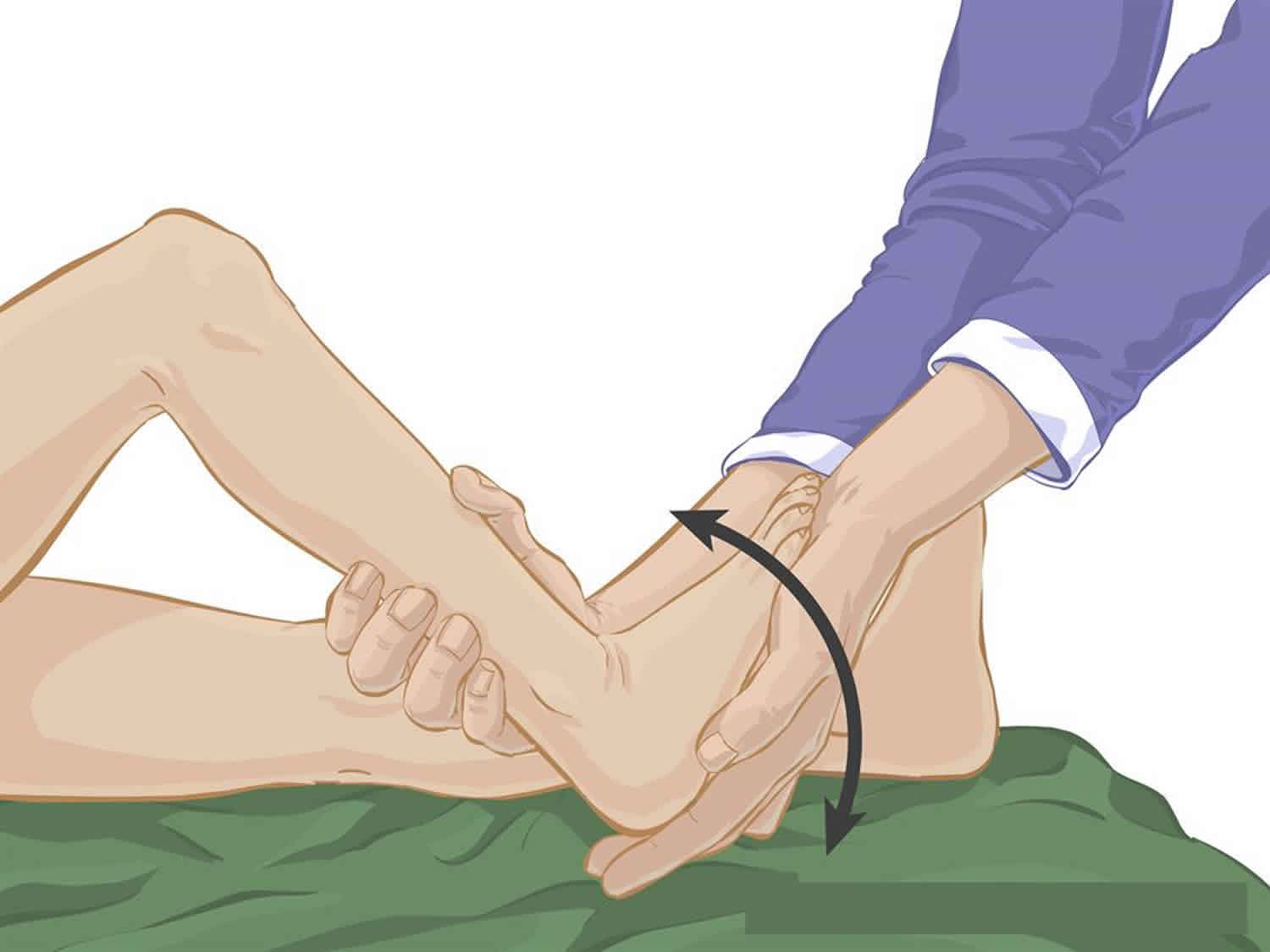
Achilles tendon stretch
- Have your child lie on their back on a comfortable surface such as a firm bed.
- With their knee bent, bring your child’s foot upwards, toward their head, bending their ankle.
- Hold the stretch at the end of the movement (that is, as far as your child’s range of motion will permit) for 15 to 30 seconds. This should not be painful for your child.
- Bring your child’s foot back to a normal position. Repeat the exercise 10 times on each leg, daily.
Figure 3. Achilles tendon stretch
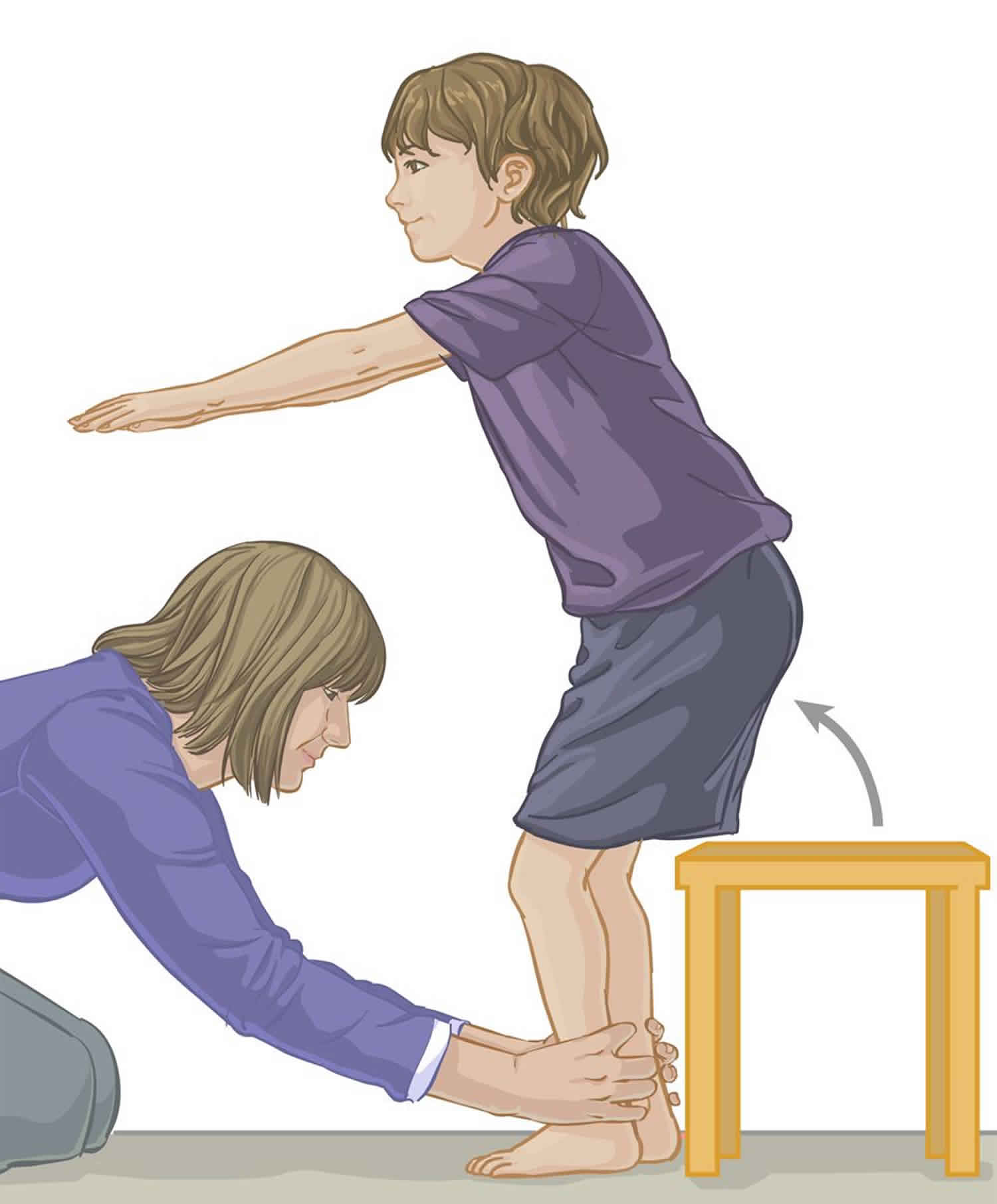
Sit to stand
- Have your child sit on a children’s sized chair or stool.
- Place your hands below their knees, providing a moderate, constant pressure downwards as a cue to keep their heels on the floor.
- Have your child practice standing up while keeping their heels on the ground.
- Make this exercise fun by playing a game of high five, blowing bubbles, reaching for objects, working in front of a mirror or singing songs.
Figure 4. Sit to stand
Exercises suitable for children ages six years and up
Calf Stretch
- Have your child stand approximately two feet from a wall. Place both of their hands at shoulder height against the wall.
- With their right knee straight, have them step towards the wall with the left foot. They should lean in until a stretch is felt in the back of the right calf. Make sure they keep the heel of the right foot on the ground.
- Hold the stretch for 15 to 30 seconds.
- Repeat the exercise 10 times on each leg, daily.
Figure 5. Calf stretch

Other exercises include:
- Marching on the spot. Have your child bring their knees up high and then land with a flat foot.
- Walking uphill.
- Walking on uneven surfaces such as in a playground or sand.
- Walking on the heels only. Keep the toes off the ground at all times.
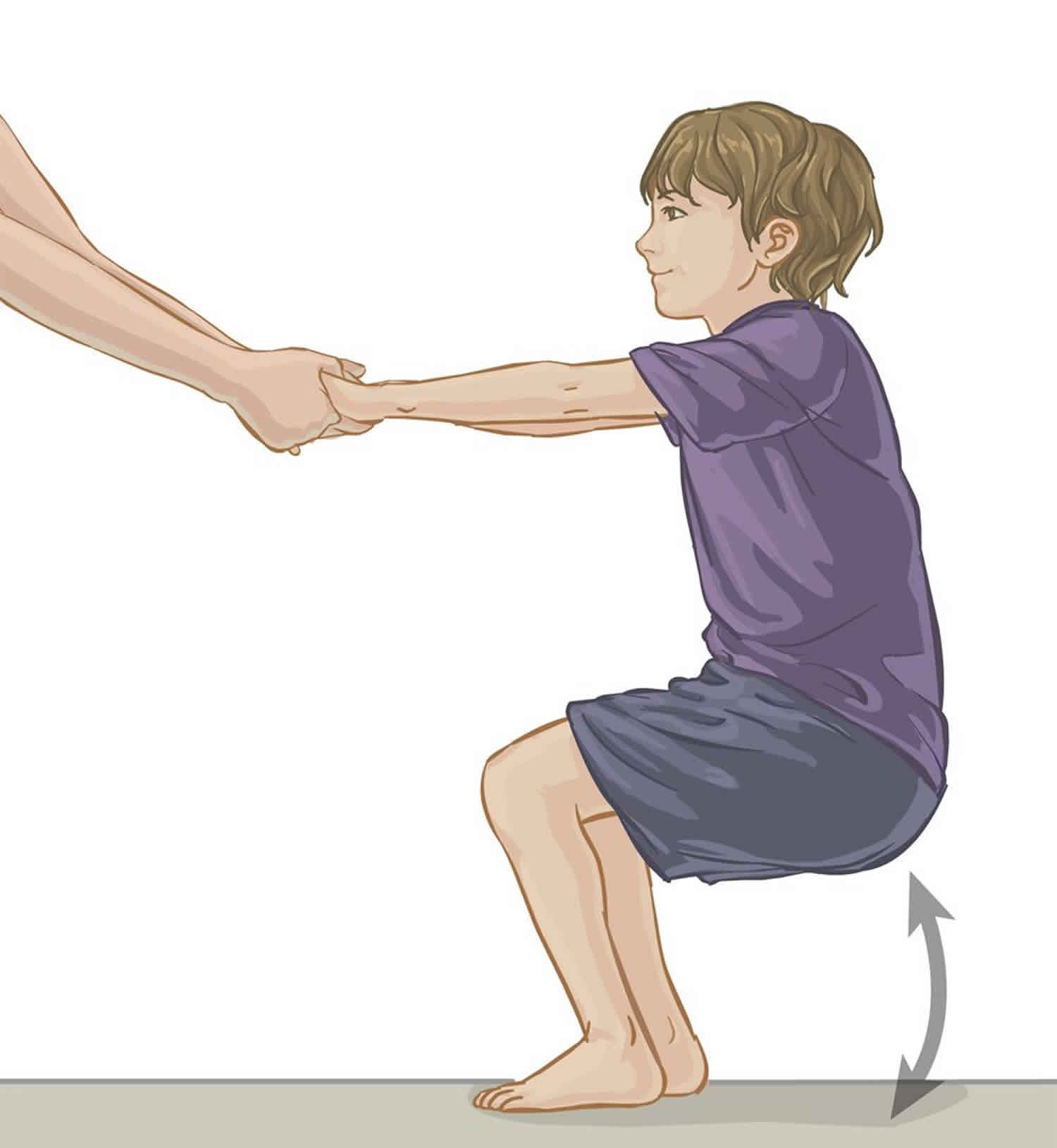
- Practicing squats. With feet flat on the floor, hip width apart, have your child slowly lower their body all the way to the floor by bending at their knees and hips but keeping their chest upright.
Figure 6. Squats
Surgical treatment
Toe-walking children over the age of 5, the calf muscles and Achilles tendons may be so tight that walking flat-footed is not possible. For these patients, the doctor may recommend a surgical procedure to lengthen the Achilles tendons. Lengthening the tendons will improve range of motion and allow better function of the foot and ankle.
The specific part of the tendon that is lengthened depends on whether or not the patient’s foot can be positioned flat at the ankle with his or her knee bent. There are several techniques used to lengthen different areas of the tendon. Your doctor will talk with you about which technique is best for your child.
The procedure is usually done on an outpatient basis (no overnight stay). After the tendons are lengthened, while your child is still asleep, your doctor will place his or her legs in short leg walking casts. These are typically worn for 4 to 6 weeks.
Recovery
Physical therapy is usually recommended after both surgical and nonsurgical treatment to help the patient learn to walk flat-footed more consistently. Physical therapy after surgery typically does not begin until the walking casts have been removed.
Toe walking prognosis
Most patients improve over time and are able to participate in normal activities and sports. However, studies show that some children will continue to toe walk—even after serial casting or surgery.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}