



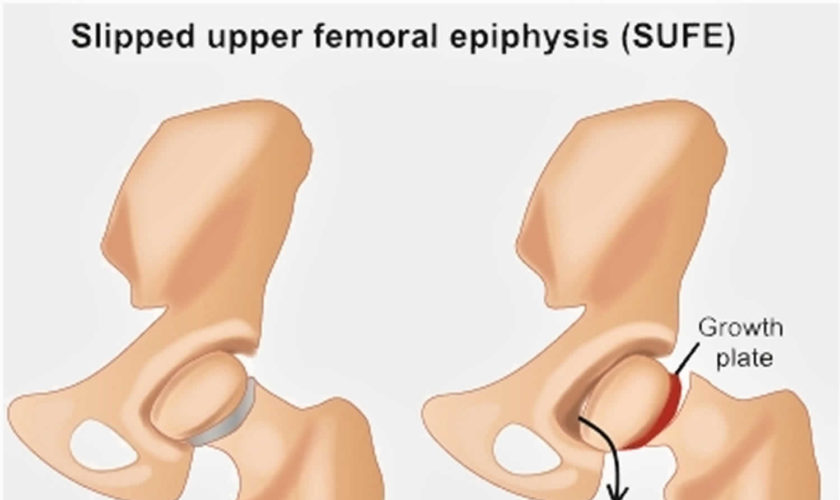
Slipped capital femoral epiphysis
Slipped capital femoral epiphysis (SCFE) also called slipped upper femoral epiphysis (SUFE), is one of the most common developmental conditions of the hip joint that occurs in teens and pre-teens who are still growing 1). For reasons that are not well understood, in slipped capital femoral epiphysis (slipped upper femoral epiphysis), a weakness of the growth plate (physis, the area at the end of the bone responsible for bone growth) in the upper end of the thigh bone (femur) causes the head, or “ball,” of the thigh bone (femoral head, epiphysis) to slip off downwards and backwards off the neck of the thigh bone, much as a scoop of ice cream can slip off the top of a cone. This causes pain, stiffness, and instability in the affected hip. Slipped capital femoral epiphysis usually develops gradually over time and is more common in boys than girls. Slipped capital femoral epiphysis is usually an emergency and must be diagnosed and treated early.
Slipped capital femoral epiphysis usually develops during periods of rapid growth, shortly after the onset of puberty. In boys, this most commonly occurs between the ages of 12 and 16; in girls, between the ages of 10 and 14.
Sometimes slipped capital femoral epiphysis occurs suddenly after a minor fall or trauma. More often, however, the condition develops gradually over several weeks or months, with no previous injury.
Slipped capital femoral epiphysis usually occurs on only one side; however, in up to 40 percent of patients (particularly those younger than age 10) slipped capital femoral epiphysis will occur on the opposite side, as well—usually within 18 months. If only one hip is affected, the other hip will eventually slip 30 to 60 percent of the time.
A slipped capital femoral epiphysis is actually a fracture of the growth plate. The fracture is usually a fairly stable one, and the slippage occurs very slowly. Occasionally, the gradual slippage can become very unstable and the ball can completely slip, leading to severe deformity and even blood supply problems to the “ball.” For this reason, every hip with slipped capital femoral epiphysis should be treated immediately to prevent unstable slipped capital femoral epiphysis.
The signs and symptoms of slipped capital femoral epiphysis (SCFE) include:
- Pain in the groin, thigh, or knee. Knee pain may be the only symptom present and can lead to a delayed diagnosis as there is no problem with the knee. The pain in the knee is a ‘referred pain’ from the hip joint.
- A limp or a turned out leg are usually present. The limp may be painless, but there is usually pain associated with the limp.
- Leg length discrepancy: The affected leg may also appear shorter than the unaffected side.
- Pain in the hip that’s aggravated by activity and that may subside with rest
- Walking with a limp, trouble walking, or feeling like the leg is “giving way”
- Walking with a leg turned outward (unilateral slip)
- Walking with a waddle (bilateral slip)
- Inability to sit with knees straight ahead (knees tend to turn outward)
Slipped capital femoral epiphysis (SCFE) is rare condition that is slightly more likely to occur in boys than girls. SCFE occurs in about one per 1,000 to one per 10,000 children and teens; children ages 12 to 14 years are most at risk. SCFE is more prevalent in the northeast region of the U.S. than in the southwest and is more prevalent among African-Americans.
Treatment for slipped capital femoral epiphysis involves surgery to stop the head of the femur from slipping any further. To achieve the best outcome, it is important to be diagnosed as quickly as possible. Without early detection and proper treatment, slipped capital femoral epiphysis can lead to potentially serious complications, including painful arthritis in the hip joint.
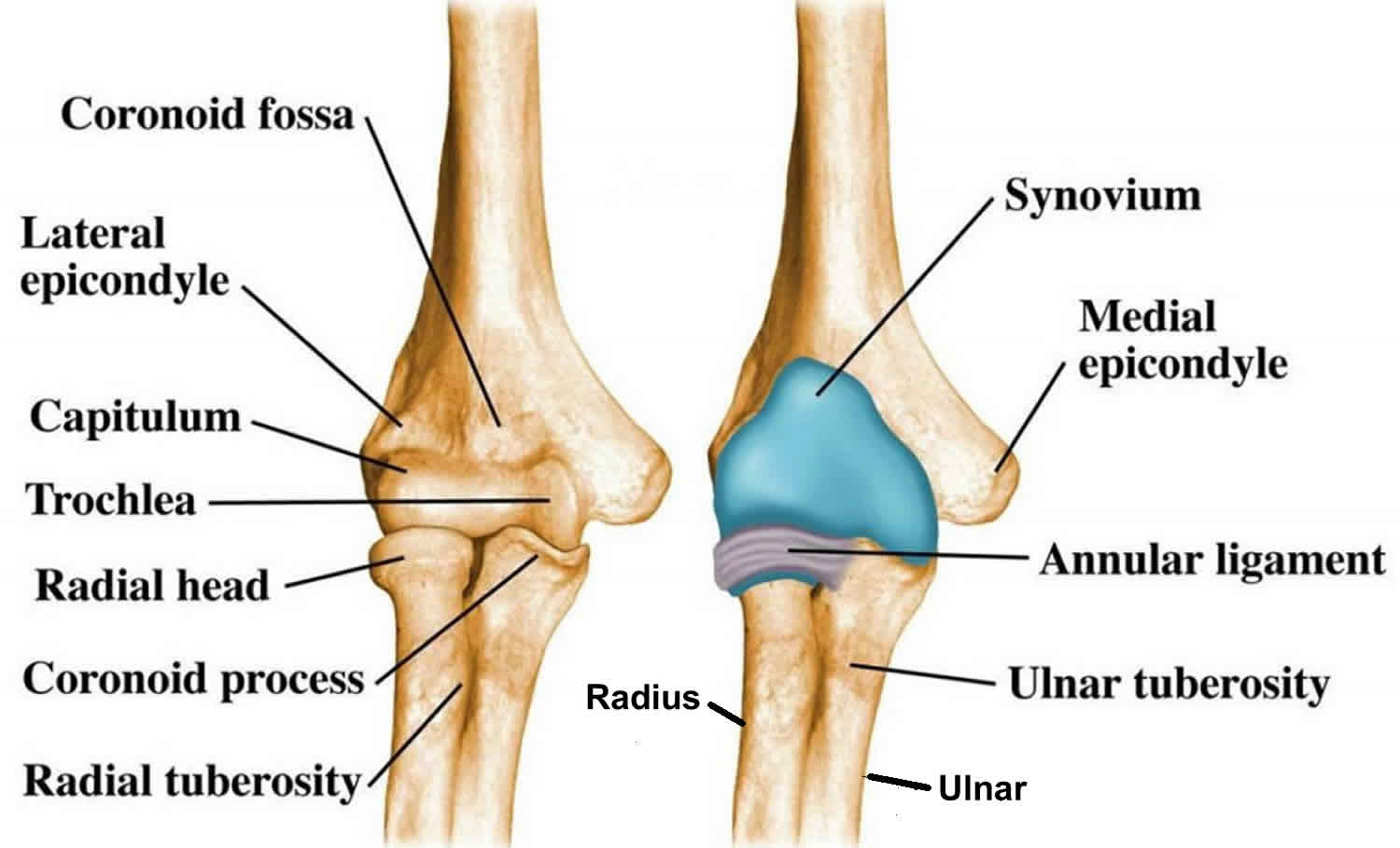
Hip anatomy
The hip is a ball-and-socket joint. The socket is formed by the acetabulum, which is part of the large pelvis bone. The ball is the femoral head, which is the upper end of the femur (thighbone).
Like the other long bones in the body, the femur does not grow from the center outward. Instead, growth occurs at each end of the bone around an area of developing cartilage called the growth plate (physis).
Growth plates are located between the widened part of the shaft of the bone (metaphysis) and the end of the bone (epiphysis). The epiphysis at the upper end of the femur is the growth center that eventually becomes the femoral head.
Figure 1. Normal anatomy of the hip
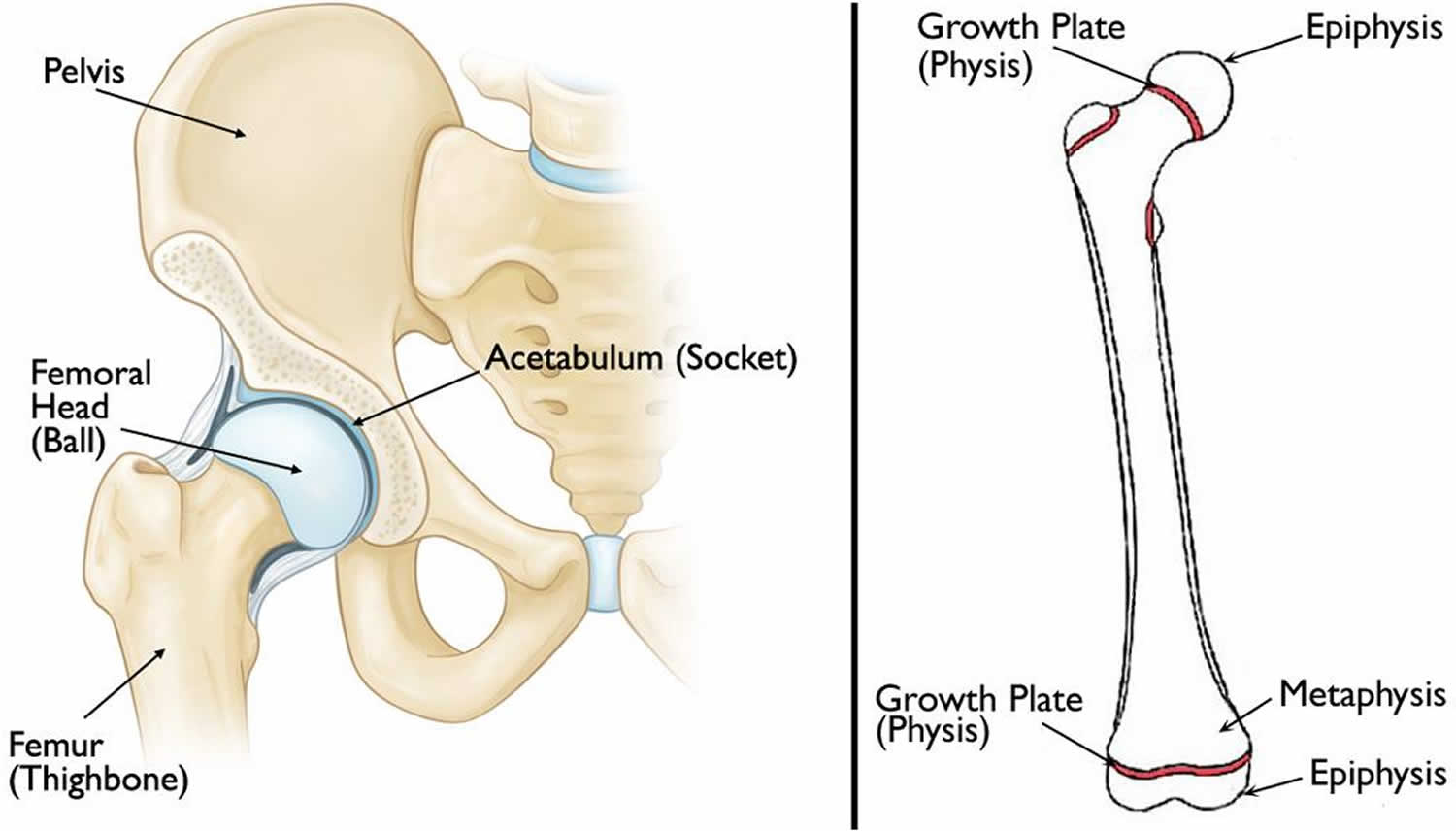
Footnote: (Left) Normal anatomy of the hip. (Right) The location of the growth plates and epiphyses at the ends of the femur (thighbone). The epiphysis at the upper end of the bone eventually becomes the femoral head.
Figure 2. Slipped capital femoral epiphysis (SCFE)
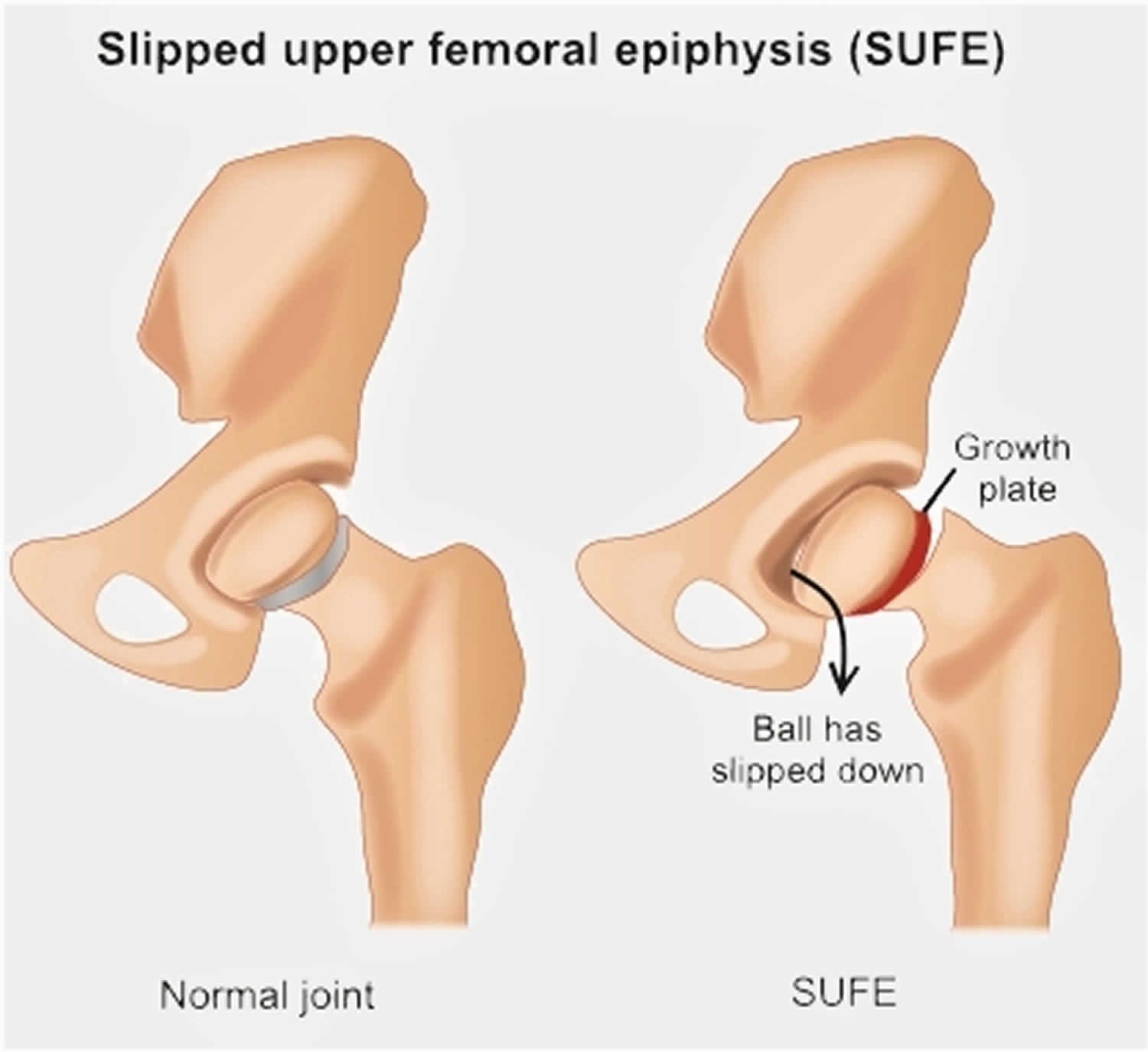
Slipped capital femoral epiphysis types
Slipped capital femoral epiphysis is often described based on whether the patient is able to bear weight on the affected hip. Knowing the type of slipped capital femoral epiphysis will help your doctor determine treatment.
Types of slipped capital femoral epiphysis include:
- Stable slipped capital femoral epiphysis. In stable slipped capital femoral epiphysis, the patient is able to walk or bear weight on the affected hip, either with or without crutches. Most cases of slipped capital femoral epiphysis are stable slips.
- Unstable slipped capital femoral epiphysis. This is a more severe slip. The patient cannot walk or bear weight, even with crutches. Unstable slipped capital femoral epiphysis requires urgent treatment. Complications associated with slipped capital femoral epiphysis are much more common in patients with unstable slips.
Slipped capital femoral epiphysis causes
The cause of slipped capital femoral epiphysis is not known. Slipped capital femoral epiphysis is more likely to occur during a growth spurt and is more common in boys than girls.
Risk factors that make someone more likely to develop slipped capital femoral epiphysis include:
- Excessive weight or obesity—most patients are above the 95th percentile for weight. This increases the stress across the growth plate of the hip bone which is the site where the slip occurs.
- Family history of slipped capital femoral epiphysis
- Trauma: Occasionally a child may have a fall at the at the time the problem started, however there may have been an underlying degree of SUFE that was aggravated by the fall.
- An endocrine or metabolic disorder, such as hypothyroidism, growth hormone deficiency, and hypogonadism—this is more likely to be a factor for patients who are older or younger than the typical age range for slipped capital femoral epiphysis (10 to 16 years of age). Slipped capital femoral epiphysis develops during puberty, a time of many hormonal changes. Rapid growth occurs in response to increased levels of growth hormone. This rapid growth is associated with an increased size in the growth plate of the hip bone and this may contribute to the decreased strength seen at puberty.
- Bone problems related to kidney disease
- Treatments for disease, like radiation and chemotherapy for cancer or taking certain medicines, including steroids
Slipped capital femoral epiphysis symptoms
Symptoms of slipped capital femoral epiphysis vary, depending upon the severity of the condition.
A patient with mild or stable slipped capital femoral epiphysis will usually have intermittent pain in the groin, hip, knee and/or thigh for several weeks or months. This pain usually worsens with activity. The patient may walk or run with a limp after a period of activity.
In more severe or unstable slipped capital femoral epiphysis, symptoms may include:
- Sudden onset of pain, often after a fall or injury
- Inability to walk or bear weight on the affected leg
- Outward turning (external rotation) of the affected leg
- Discrepancy in leg length—the affected leg may appear shorter than the opposite leg
Slipped capital femoral epiphysis complications
Although early detection and proper treatment of slipped capital femoral epiphysis will help decrease the chance of complications, some patients will still experience problems.
The most common complications following slipped capital femoral epiphysis are avascular necrosis and chondrolysis.
Avascular necrosis (osteonecrosis)
If severe cases, slipped capital femoral epiphysis causes the blood supply to the femoral head to become limited. This can lead to a gradual and very painful collapse of the bone, a condition called avascular necrosis or osteonecrosis. Avascular necrosis of the femoral head is thought to result from vascular damage during the time of the initial traumatic event, but it may result from forceful reduction during the time of surgery. The amount of energy, magnitude of epiphyseal damage and displacement, level of increased intra-articular pressure, and degree of vascular occlusion have been implicated in this process. The risk of avascular necrosis is up to 47% with an unstable slipped capital femoral epiphysis.
When the bone collapses, the articular cartilage covering the bone also collapses. Without this smooth cartilage, bone rubs against bone, leading to painful arthritis in the joint. For some patients with avascular necrosis, further surgery may be needed to reconstruct the hip.
Avascular necrosis is more likely to occur in patients with unstable slipped capital femoral epiphysis. Because evidence of osteonecrosis may not be seen on x-ray for up to 12 months following surgery, the patient will be monitored with x-rays during this period of time.
Treatment options are limited (eg, bone grafting, osteotomy to change the position of the femoral head), but often these patients will eventually need a total hip replacement.
Chondrolysis
Chondrolysis is a rare but serious complication of slipped capital femoral epiphysis. In chondrolysis, the articular cartilage on the surface of the hip joint degenerates very rapidly, leading to pain, deformity, and permanent loss of motion in the affected hip.
Although the cause of chondrolysis is not yet fully understood by doctors, it is believed that it may result from inflammation in the hip joint.
Aggressive physical therapy and anti-inflammatory medications may be prescribed for patients who develop chondrolysis. Over time, there may be some gradual return of motion in the hip.
Osteoarthritis
Osteoarthritis is a late complication. There is evidence that increased risk of early degenerative change may result from avascular necrosis, chondrolysis, or alterations of the hip biomechanics following slippage. In general, the more severe the deformity and/or slipped capital femoral epiphysis, the higher risk of developing arthritis. Mild deformities may have few consequences.
Leg-length inequality
Leg-length inequality may result from incomplete reduction, avascular necrosis, chondrolysis, or secondary coxa vara.
Hardware failure and “outgrowing” hardware may cause loss of fixation. Although rare, postoperative infection may occur.
Slipped capital femoral epiphysis diagnosis
During the examination, your doctor will ask about your child’s general health and medical history. He or she will then talk with you about your child’s symptoms and ask when the symptoms began.
While your child is lying down, the doctor will perform a careful examination of the affected hip and leg, looking for:
- Pain with extremes of motion
- Limited range of motion in the hip–especially limited internal rotation
- Involuntary muscle guarding and muscle spasms
Your doctor will also observe your child’s gait (the way he or she walks). A child with slipped capital femoral epiphysis may limp or have an abnormal gait.
X-rays
This type of study provides images of dense structures, such as bone. Your doctor will order x-rays of the pelvis, hip, and thigh from several different angles to help confirm the diagnosis.
In a patient with slipped capital femoral epiphysis, an x-ray will show that the head of the thighbone appears to be slipping off the neck of the bone.
An MRI or CT scan is rarely required.
Slipped capital femoral epiphysis treatment
Treatment of slipped capital femoral epiphysis requires surgery to correct the problem. The aim of surgery is to stop the hip bone from slipping further and make sure any slip that has occurred is corrected. The earlier the treatment, the better the outcomes and for this reason a quick diagnosis is very important. The treatment for a stable slipped capital femoral epiphysis (when the child can walk on the affected side) is slightly different to the treatment of unstable slipped capital femoral epiphysis (when the child is unable to walk on the affected side).
Early diagnosis of slipped capital femoral epiphysis provides the best chance of stabilizing the hip and avoiding complications. When treated early and appropriately, long-term hip function can be expected to be very good.
Once slipped capital femoral epiphysis is confirmed, your child will not be allowed to bear weight on his or her hip and will probably be admitted to the hospital. In most cases, surgery is performed within 24 to 48 hours.
Stable slipped capital femoral epiphysis
The treatment for stable slipped capital femoral epiphysis is usually to insert a single screw into the the thigh bone across the place where the bone is slipping. This holds the bone together and prevents any further slip. In some cases, the surgeon may decide to perform operations that include grafting bone or changing the angle of the bone but usually a single screw is all that is needed.
After the operation, crutches are recommended for several days. After this, the child is recovered to walk on the affected side to ensure the bone heals normally.
Unstable slipped capital femoral epiphysis
Usually, unstable slipped capital femoral epiphysis is considered a more urgent condition and operations will usually be carried out within a short time of the diagnosis. Before the operation, the joint may be drained of any fluid that may have accumulated due to the inflammation caused by the slip. The operation involves first lining the bones up by the use of traction and then placing one or two screws across the place where the bone has slipped.
Following the operation, crutches must be used for 6-8 weeks in order to prevent any further slip. Physiotherapy rehabilitation may be required to ensure the hip is moving well and that the surrounding muscles are strong.
SCFE surgery
The surgical procedure your doctor recommends will depend upon the severity of the slip. Procedures used to treat slipped capital femoral epiphysis include:
In situ fixation
This is the procedure used most often for patients with stable or mild slipped capital femoral epiphysis. The doctor makes a small incision near the hip, then inserts a metal screw across the growth plate to maintain the position of the femoral head and prevent any further slippage.
Over time, the growth plate will close, or fuse. Once the growth plate is closed, no further slippage can occur.
Open reduction
In patients with unstable slipped capital femoral epiphysis, the doctor may first make an open incision in the hip, then gently manipulate (reduce) the head of the femur back into its normal anatomic position.
The doctor will then insert one or two metal screws to hold the bone in place until the growth plate closes. This is a more extensive procedure and requires a longer recovery time.
In situ fixation in the opposite hip
Some patients are at higher risk for slipped capital femoral epiphysis occurring on the opposite side. If this is the case with your child, your doctor may recommend inserting a screw into his or her unaffected hip at the same time to reduce the risk of slipped capital femoral epiphysis. Your doctor will talk with you about whether this is appropriate for your child.
Recovery
Weight bearing
After surgery, your child will be on crutches with protected weight bearing for 6-8 weeks. Your child’s doctor will give you specific instructions about when full weight bearing can begin. To prevent further injury, it is important to closely follow your doctor’s instructions.
Physical therapy
A physical therapist will provide specific exercises to help strengthen the hip and leg muscles and improve range of motion. Physical therapy for strengthening, proprioception, balance, and endurance training may be helpful. Most children can then return to full activity once they are pain free with full strength. However, some literature advocates for not allowing a return to contact sports until the physis has closed.
Sports and other activities
For a period of time after surgery, your child will be restricted from participating in vigorous sports and activities. This will help minimize the chance of complications and enable healing to take place. Your doctor will tell you when your child can safely resume his or her normal activities.
Follow-up care
Your child will return to the doctor for follow-up visits for 18 to 24 months after surgery. These visits may include x-rays every 3 to 4 months to ensure that the growth plate has closed and that no complications have developed.
Depending upon the patient’s age and other factors, a team approach that includes a general pediatrician, endocrinologist, and/or dietician may be necessary for comprehensive care in the long run.
Slipped capital femoral epiphysis prognosis
Most patients with slipped capital femoral epiphysis who are treated with urgent in situ fixation do well. However, in those cases with severe slippage and resultant deformity, long-term sequelae may result (eg, avascular necrosis, chondrolysis, leg-length discrepancy, stiffness, osteoarthritis). Although conservative modalities (eg, therapy, analgesics, orthotics, assistive aids) are used initially for symptomatic relief, urgent operative intervention is indicated. Young patients with unremitting pain, loss of motion, and stiffness secondary to chondrolysis, avascular necrosis, or osteoarthritis may require salvage hip arthrodeses. In hips that are incompletely damaged, proximal osteotomies may aid in redirecting the joint forces to less damaged areas of the articular femoral head.
References [ + ]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}