



What is rhabdomyosarcoma
Rhabdomyosarcoma is a cancer that starts in skeletal muscles. Rhabdomyosarcoma can occur in children or adults. Skeletal (voluntary) muscles are muscles that you control to move parts of your body. Skeletal muscle is under voluntary control and is the major muscle component of your body. Skeletal muscle is responsible for the maintenance of body posture and also for the gross and fine movements of the limbs. In addition, the skeletal muscle of the eye (the extraocular muscles) are responsible for eye movements; and the visceral striated muscle is responsible for important elements of speech, breathing and swallowing.
About 3% of all childhood cancers are rhabdomyosarcoma. Childhood rhabdomyosarcoma is a soft tissue malignant tumor of mesenchymal origin. More than 70% of malignant mesenchymal tumors are rhabdomyosarcomas and occurs with highest incidence in children and adolescents. Rhabdomyosarcoma accounts for approximately 3.5% of the cases of cancer among children aged 0 to 14 years and 2% of the cases among adolescents and young adults aged 15 to 19 years 1). The median age at presentation is 5 years. Rhabdomyosarcoma is slightly more common in boys than in girls. No particular race or ethnic group seems to have an unusually high rate of rhabdomyosarcoma. The incidence is 4.5 cases per 1 million children, which translates into about 350 new cases of rhabdomyosarcoma occur each year in the United States 2). Fifty percent of these cases are seen in the first decade of life 3). Males have a higher incidence of embryonal tumors, and blacks have a slightly higher incidence of alveolar tumors 4). The number of new cases has not changed much over the past few decades.
Incidence may depend on the histologic subtype of rhabdomyosarcoma, as follows:
- Embryonal: Patients with embryonal rhabdomyosarcoma are predominantly male (male to female ratio, 1.5). The peak incidence is in the 0- to 4-year age group, with approximately 4 cases per 1 million children, with a lower rate in adolescents, approximately 1.5 cases per 1 million adolescents. This subtype constitutes 57% of patients in the Surveillance, Epidemiology, and End Results (SEER) database 5).
- Alveolar: The incidence of alveolar rhabdomyosarcoma does not vary by sex and is constant from ages 0 to 19 years, with approximately 1 case per 1 million children and adolescents. This subtype constitutes 23% of patients in the SEER database 6).
- Other: Pleomorphic/anaplastic, mixed type, and spindle cell subtypes each constitute less than 2% of children with rhabdomyosarcoma 7).
The following are the most common primary sites for rhabdomyosarcoma 8):
- Head and neck region (parameningeal) (approximately 25%).
- Genitourinary tract (approximately 31%).
- Extremities (approximately 13%). Within extremity tumors, tumors of the hand and foot occur more often in older patients and have an alveolar histology 9).
Other less common primary sites include the trunk, chest wall, perineal/anal region, and abdomen, including the retroperitoneum and biliary tract 10).
There are 3 distinct types of rhabdomyosarcoma:
- Embryonal rhabdomyosarcoma
- Alveolar rhabdomyosarcoma
- Anaplastic rhabdomyosarcoma and undifferentiated sarcoma (formerly called pleomorphic rhabdomyosarcoma)
The response to treatment varies according to the type of rhabdomyosarcoma cancer you have.
About 7 weeks into the development of an embryo, cells called rhabdomyoblasts (which will eventually form skeletal muscles) begin to form. These are the cells that can develop into rhabdomyosarcoma. Because rhabdomyosarcoma is a cancer of embryonal cells, it is much more common in children, although it does sometimes occur in adults.
You might think of your skeletal muscles as being mainly in our arms and legs, but these skeletal muscle cancers can start nearly anywhere in your body, even in some parts of the body that don’t normally have skeletal muscle.
Common sites of rhabdomyosarcoma include:
- Head and neck (such as near the eye, inside the nasal sinuses or throat, or near the spine in the neck)
- Urinary and reproductive organs (bladder, prostate gland, or any of the female organs)
- Arms and legs
- Trunk (chest and abdomen)
Most rhabdomyosarcomas are diagnosed in children and teens, with more than half of them in children younger than 10 years old. These tumors are usually embryonal rhabdomyosarcomas and tend to develop in the head and neck area or in the genital and urinary tracts. Alveolar rhabdomyosarcoma affects all age groups and is found more often in the arms, legs, or trunk.
It is important to realize that the treatment of rhabdomyosarcoma is a very complicated process. Younger patients may suffer long-term consequences of treatments to rhabdomyosarcoma cancer.
Treatment of rhabdomyosarcoma varies depending upon the site of the lesion, but revolves around surgical, chemotherapy and radiotherapy techniques. Surgery and radiotherapy are used to establish local control of the tumor. The early dissemination of this tumor has necessitated systemic chemotherapy to prevent the development of metastatic disease, and also to assist in the maintenance of local disease control.
Continual improvements in survival have been achieved for children and adolescents with cancer 11). Between 1975 and 2010, childhood cancer mortality decreased by more than 50% 12). For rhabdomyosarcoma, the 5-year survival rate increased over the same time, from 53% to 67% for children younger than 15 years and from 30% to 51% for adolescents aged 15 to 19 years 13).
Childhood and adolescent cancer survivors require close monitoring because side effects of cancer and its therapy may persist or develop months or years after treatment.
Embryonal rhabdomyosarcoma
Embryonal rhabdomyosarcoma usually affects children in their first 5 years of life, but it is the most common type of rhabdomyosarcoma at all ages.
The cells of embryonal rhabdomyosarcoma look like the developing muscle cells of a 6- to 8-week-old embryo. Embryonal rhabdomyosarcoma tends to occur in the head and neck area, bladder, vagina, or in or around the prostate and testicles.
Two subtypes of embryonal rhabdomyosarcoma, botryoid and spindle cell rhabdomyosarcomas, tend to have a better prognosis (outlook) than the more common conventional form of embryonal rhabdomyosarcoma.
Alveolar rhabdomyosarcoma
Alveolar rhabdomyosarcoma typically affects all age groups equally. It makes up a larger portion of rhabdomyosarcoma in older children and teens than in younger children (because embryonal rhabdomyosarcoma is less common at older ages).
Alveolar rhabdomyosarcoma most often occurs in large muscles of the trunk, arms, and legs. The cells of alveolar rhabdomyosarcoma look like the normal muscle cells seen in a 10-week-old fetus.
Alveolar rhabdomyosarcoma tends to grow faster than embryonal rhabdomyosarcoma and usually requires more intense treatment.
Anaplastic rhabdomyosarcoma and undifferentiated sarcoma
Anaplastic rhabdomyosarcoma (formerly called pleomorphic rhabdomyosarcoma) is an uncommon type that occurs in adults but is very rare in children.
Some doctors also group undifferentiated sarcomas with the rhabdomyosarcomas. Using lab tests, doctors can tell that these cancers are sarcomas, but the cells don’t have any features that help classify them further.
Both of these uncommon cancers tend to grow quickly and usually require intensive treatment.
Rhabdomyosarcoma in adults
Most rhabdomyosarcomas develop in children, but they can also occur in adults. Adults are more likely to have faster-growing types of rhabdomyosarcoma and to have them in parts of the body that are harder to treat. Because of this, rhabdomyosarcoma in adults is often harder to treat effectively.
What Are the Differences Between Cancers in Adults and Children?
Cancers that develop in children are often different from the types that develop in adults. Childhood cancers are often the result of DNA changes in cells that take place very early in life, sometimes even before birth. Unlike many cancers in adults, childhood cancers are not strongly linked to lifestyle or environmental risk factors.
There are exceptions, but childhood cancers tend to respond better to treatments such as chemotherapy. Children’s bodies also tend to tolerate chemotherapy better than adults’ bodies do. But cancer treatments such as chemotherapy and radiation therapy can have long-term side effects, so children who have had cancer need careful attention for the rest of their lives.
Since the 1960s, most children and teens with cancer have been treated at specialized centers designed for them. These centers offer the advantage of being treated by a team of specialists who know the differences between adult and childhood cancers, as well as the unique needs of children with cancer and their families. This team usually includes pediatric oncologists (childhood cancer doctors), surgeons, radiation oncologists, pathologists, pediatric oncology nurses, and nurse practitioners.
These centers also have psychologists, social workers, child life specialists, nutritionists, rehabilitation and physical therapists, and educators who can support and educate the entire family.
Most children with cancer in the United States are treated at a center that is a member of the Children’s Oncology Group. All of these centers are associated with a university or children’s hospital. As we have learned more about treating childhood cancer, it has become even more important that treatment be given by experts in this area.
When a child or teen is diagnosed with cancer, it affects every family member and nearly every aspect of the family’s life.
Rhabdomyosarcoma prognosis
The prognosis (outlook) for people with rhabdomyosarcoma depends on many factors, including the type of rhabdomyosarcoma, the location and size of the tumor, the results of surgery, and whether the cancer has metastasized (spread). Children aged 1 to 9 tend to have a better outlook than infants or older children or adults.
Before the advent of chemotherapy in the 1970s the outlook for patients with rhabdomyosarcoma was universally poor. A 5 year survival rate was less than 20%. More recently, however, the cure rate for rhabdomyosarcoma has risen to approximately 70% following the introduction of postoperative systemic chemotherapy.
Parameningeal head and neck tumors and intra-abdominal tumors have the worst prognosis, whilst those in the orbit and urogenital tract are associated with a good prognosis. Female patients have a worse prognosis than do males.
Prognostic factors
Rhabdomyosarcoma is usually curable in children with localized disease who receive combined-modality therapy, with more than 70% of patients surviving 5 years after diagnosis 14). Relapses are uncommon in patients who were alive and event free at 5 years, with a 10-year late-event rate of 9%. Relapses are more common, however, in patients who have unresectable disease in an unfavorable site at diagnosis and in patients who have metastatic disease at diagnosis 15).
The prognosis for a child or adolescent with rhabdomyosarcoma is related to the following clinical and biological factors:
- Age.
- Site of origin.
- Tumor size.
- Resectability.
- Histopathologic subtype.
- PAX3/PAX7-FOXO1 gene fusion status.
- Metastases at diagnosis.
- Lymph node involvement at diagnosis.
- Biological characteristics.
- Response to therapy.
Because treatment and prognosis partly depend on the histology and molecular genetics of the tumor, it is necessary that the tumor tissue be reviewed by pathologists and cytogeneticists/molecular geneticists with experience in the evaluation and diagnosis of tumors in children. Additionally, the diversity of primary sites, the distinctive surgical and radiation therapy treatments for each primary site, and the subsequent site-specific rehabilitation underscore the importance of treating children with rhabdomyosarcoma in medical centers with appropriate experience in all therapeutic modalities.
Age
Children aged 1 to 9 years have the best prognosis, while those younger and older fare less well. In recent Intergroup Rhabdomyosarcoma Study Group (IRSG) trials 16), the 5-year failure-free survival rate was 57% for patients younger than 1 year, 81% for patients aged 1 to 9 years, and 68% for patients older than 10 years. Five-year survival rates were 76% for patients younger than one year, 87% for patients aged 1 to 9 years, and 76% for patients older than 10 years. Historical data show that adults fare less well than children (5-year overall survival [overall survival] rates, 27% ± 1.4% and 61% ± 1.4%, respectively) 17).
Young age
Infants may do poorly because chemotherapy doses are reduced by 50% on the basis of reports that they have higher death rates related to chemotherapy toxicity when compared with older patients; therefore, young patients may be underdosed 18). In addition, infants younger than 1 year are less likely to receive radiation therapy for local control, because of concern about the high incidence of late effects in this age group 19).
The 5-year failure-free survival rate for infants was found to be 67%, compared with 81% in a matched group of older patients treated by the Children’s Oncology Group 20). This inferior failure-free survival rate was largely because of a relatively high rate of local failure.
In another retrospective study of 126 patients (aged ≤24 months) who were enrolled on the ARST0331 (NCT00075582) (https://clinicaltrials.gov/ct2/show/NCT00075582) and ARST0531 (NCT00354835) (https://clinicaltrials.gov/ct2/show/NCT00354835) trials, the 5-year local failure rate was 24%, the 5-year event-free survival (event-free survival) rate was 68.3%, and the overall survival rate was 81.9%. Forty-three percent of the patients had an individualized local therapy plan that more frequently omitted radiation therapy. These patients had inferior local control and event-free survival rates 21).
Members of the Cooperative Weichteilsarkom Studiengruppe reviewed 155 patients with rhabdomyosarcoma presenting from birth to age 12 months; 144 patients had localized disease; 11 patients had metastases; 32 patients presented with alveolar rhabdomyosarcoma pathology. The following results were reported 22):
- Of the 144 patients with localized disease, 129 patients had a complete response.
- Fifty-one infants had a recurrence of their disease; 63% of patients with alveolar rhabdomyosarcoma relapsed, and 28% of patients with embryonal rhabdomyosarcoma relapsed.
- Five-year overall survival rates were 69% for patients with localized disease, 14% for patients with metastatic disease, and 41% for patients with relapsed disease.
Older children
In older children, the upper dosage limits of vincristine and dactinomycin are based on body surface area and these patients may require reduced vincristine doses because of neurotoxicity 23).
Adolescents
A report from the Associazione Italiana Ematologia Oncologia Paediatrica (AIEOP) Soft Tissue Sarcoma Committee 24) suggests that adolescents may have more frequent unfavorable tumor characteristics, including alveolar histology, regional lymph node involvement, and metastatic disease at diagnosis, accounting for their poor prognosis. This study also found that 5-year overall survival and progression-free survival rates were somewhat lower in adolescents than in children, but the differences among age groups younger than 1 year and aged 10 to 19 years at diagnosis were significantly worse than those in the group aged 1 to 9 years 25).
Adults
Adult patients with rhabdomyosarcoma have a higher incidence of pleomorphic histology (19%) than do children (<2%). Adults also have a higher incidence of tumors in unfavorable sites than do children 26).
Site of origin
Prognosis for childhood rhabdomyosarcoma varies according to the primary tumor site (see Table 1).
Table 1. 5-Year Survival by Primary Site of Disease
| Primary Site | Number of Patients | Survival at 5 Years (%) |
|---|---|---|
| a) Patients treated on Intergroup Rhabdomyosarcoma Study III 27). | ||
| b) Patients treated on Intergroup Rhabdomyosarcoma Studies I–IV 28). | ||
| Orbit a | 107 | 95 |
| Superficial head and neck (nonparameningeal) a | 106 | 78 |
| Cranial parameningeal a | 134 | 74 |
| Genitourinary (excluding bladder/prostate) a | 158 | 89 |
| Bladder/prostate a | 104 | 81 |
| Extremity a | 156 | 74 |
| Trunk, abdomen, perineum, etc. a | 147 | 67 |
| Biliary b | 25 | 78 |
Tumor size
Children with tumors 5 cm or less have improved survival compared with children with tumors larger than 5 cm 29). Both tumor volume and maximum tumor diameter are associated with outcome 30).
A retrospective review of soft tissue sarcomas in children and adolescents suggests that the 5 cm cutoff used for adults with soft tissue sarcoma may not be ideal for smaller children, especially infants. The review identified an interaction between tumor diameter and body surface area 31). This was not confirmed by a Children’s Oncology Group study of patients with intermediate-risk rhabdomyosarcoma 32). This relationship requires prospective study to determine the therapeutic implications of the observation.
Resectability
The extent of disease after the primary surgical procedure (i.e., the Surgical-pathologic Group, also called the Clinical Group) is correlated with outcome 33). In the IRS-III study, patients with localized, gross residual disease after initial surgery (Surgical-pathologic Group III) had a 5-year survival rate of approximately 70%, compared with a rate of more than 90% for patients without residual tumor after surgery (Group I) and a rate of approximately 80% for patients with microscopic residual tumor after surgery (Group II) 34). Group I and Group II represent a minority of patients; approximately 50% of patients have unresectable Group III disease at time of diagnosis 35).
Resectability without functional impairment is related to initial size and site of the tumor and does not account for the biology of the disease. Outcome is optimized with the use of multimodality therapy. All patients require chemotherapy, and at least 85% of patients also benefit from radiation therapy, with favorable outcomes even for patients with nonresectable disease. In the IRS-IV study, the Group III patients with localized unresectable disease who were treated with chemotherapy and radiation therapy had a 5-year failure-free survival rate of about 75% and a local control rate of 87% 36).
Histopathologic subtype
The alveolar subtype is more prevalent among patients with less favorable clinical features (e.g., younger than 1 year or older than 10 years, extremity and truncal primary tumors, and metastatic disease at diagnosis), and is generally associated with a worse outcome than in similar patients with embryonal rhabdomyosarcoma.
- In the IRS-I and IRS-II studies, the alveolar subtype was associated with a less favorable outcome even in patients whose primary tumor was completely resected (Group I) 37).
- A statistically significant difference in 5-year survival by histopathologic subtype (82% for embryonal rhabdomyosarcoma vs. 65% for alveolar rhabdomyosarcoma) was not noted when 1,258
- IRS-III and IRS-IV patients with rhabdomyosarcoma were analyzed 38).
- In the IRS-III study, outcome for patients with Group I alveolar subtype tumors was similar to that for other patients with Group I tumors, but the alveolar patients received more intensive therapy 39).
- Patients with alveolar rhabdomyosarcoma who have regional lymph node involvement have significantly worse outcomes (5-year FFS, 43%) than patients who do not have regional lymph node involvement (5-year failure-free survival, 73%) 40).
Anaplasia has been observed in 13% of embryonal rhabdomyosarcoma cases and its presence may adversely influence clinical outcome in patients with intermediate-risk disease. However, anaplasia was not shown to be an independent prognostic variable in a multivariate analysis 41).
PAX3/PAX7-FOXO1 gene fusion status
Occasionally, patients with histology consistent with alveolar rhabdomyosarcoma do not have one of the two gene fusions that are characteristic of the disease. Patients with translocation-negative alveolar rhabdomyosarcoma have outcomes similar to those for patients with embryonal rhabdomyosarcoma and fare better than patients with fusion-positive alveolar rhabdomyosarcoma 42). For example, in a study from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group of 434 cases of intermediate-risk rhabdomyosarcoma, fusion-positive patients had a lower EFS rate (PAX3, 54% and PAX7, 65%) than did those with embryonal rhabdomyosarcoma (event-free survival rate, 77%).
In a Children’s Oncology Group study, patients with Stage 2 or 3, Group III PAX3-positive tumors had worse overall survival rates than did those with PAX7 tumors 43). Comparable results were observed in another study; patients with PAX7-positive tumors and patients with fusion-negative tumors had similar outcomes 44).
These studies also demonstrated that fusion status was a better predictor of outcome than was histology and this variable has now been incorporated into the risk stratification of patients in the current Children’s Oncology Group ARST1431 (NCT02567435) (https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCT02567435) study for patients with intermediate-risk rhabdomyosarcoma. Similar conclusions were reached in a retrospective study of three consecutive trials in the United Kingdom. The authors underscored the probable value of treating fusion-negative patients whose tumors have alveolar histology with therapy that is stage appropriate for embryonal histology tumors 45).
Metastases at diagnosis
Children with metastatic disease at diagnosis have the worst prognosis.
The prognostic significance of metastatic disease is modified by the following:
- Tumor histology (embryonal rhabdomyosarcoma is more favorable than alveolar). Only patients with alveolar histology and regional node disease have a worse prognosis provided that the regional disease is treated with radiation therapy 46).
- Age at diagnosis (<10 years for children with embryonal rhabdomyosarcoma).
- The site of metastatic disease. Patients with metastatic genitourinary (nonbladder, nonprostate) primary tumors have a more favorable outcome than do patients with metastatic disease from other primary sites 47).
- The number of metastatic sites 48).
The Children’s Oncology Group performed a retrospective review of patients enrolled on high-risk protocols for rhabdomyosarcoma. PAX fusion status correlated with clinical characteristics at diagnosis, including age, stage, histology, and extent of metastatic disease (Oberlin status). Among patients with metastatic disease, PAX-FOXO1 fusion status was not an independent predictor of outcome 49).
Lymph node involvement at diagnosis
Lymph node involvement at diagnosis is associated with an inferior prognosis 50) and clinical and/or imaging evaluation is performed before treatment and preoperatively. Sentinel lymph node identification by appropriate methodology can aid in this evaluation. Suspicious nodes are sampled surgically with open biopsy preferred to needle aspiration, although this may occasionally be appropriate. Pathologic evaluation of clinically uninvolved nodes is site specific; in the United States, it is performed for extremity sites or for boys older than 10 years with paratesticular primaries.
Data on the frequency of lymph node involvement in various sites are useful for making clinical decisions. For example, up to 40% of patients with rhabdomyosarcoma in genitourinary sites have lymph node involvement, while patients with certain head and neck sites have a much lower likelihood (<10%). Patients with nongenitourinary pelvic sites (e.g. anus/perineum) have an intermediate frequency of lymph node involvement 51).
In the extremities and select truncal sites, sentinel lymph node evaluation is a more accurate form of diagnosis than is random regional lymph node sampling. In clinically negative lymph nodes of the extremity or trunk, sentinel lymph node biopsy is the preferred form of node sampling by the Children’s Oncology Group. Technical considerations are obtained from surgical experts. Needle or open biopsy of clinically enlarged nodes is appropriate 52).
Radiation therapy is administered to patients with lymph node involvement in order to enhance regional control.
Biological characteristics
The embryonal and alveolar histologies have distinctive molecular characteristics that have been used for diagnostic confirmation, and may be useful for assigning risk group, determining therapy, and monitoring residual disease during treatment 53).
- Embryonal histology: Embryonal tumors often show loss of heterozygosity at 11p15 and gains on chromosome 8 54). Embryonal tumors have a higher background mutation rate and a higher single-nucleotide variant rate than do alveolar tumors, and the number of somatic mutations increases with older age at diagnosis 55). Genes with recurring mutations include those in the RAS pathway (e.g., NRAS, KRAS, HRAS, and NF1), which together are observed in approximately one-third of cases. Other genes with recurring mutations include FGFR4, PIK3CA, CTNNB1, FBXW7, and BCOR, all of which are present in fewer than 10% of cases 56). Embryonal histology with anaplasia: Anaplasia has been reported in a minority of children with rhabdomyosarcoma, primarily arising in children with the embryonal subtype who are younger than 10 years 57). Rhabdomyosarcoma with nonalveolar anaplastic morphology may be a presenting feature for children with Li-Fraumeni syndrome and germline TP53 mutations 58). Among eight consecutively presenting children with rhabdomyosarcoma and TP53 germline mutations, all showed anaplastic morphology. Among an additional seven children with anaplastic rhabdomyosarcoma and unknown TP53 germline mutation status, three of the seven children had functionally relevant TP53 germline mutations. The median age at diagnosis of the 11 children with TP53 germline mutation status was 40 months (range, 19–67 months).
- Alveolar histology: About 70% to 80% of alveolar tumors are characterized by translocations between the FOXO1 gene on chromosome 13 and either the PAX3 gene on chromosome 2 (t(2;13)(q35;q14)) or the PAX7 gene on chromosome 1 (t(1;13)(p36;q14)) 59). Other rare fusions include PAX3-NCOA1 and PAX3-INO80D 60). Translocations involving the PAX3 gene occur in approximately 59% of alveolar rhabdomyosarcoma cases, while the PAX7 gene appears to be involved in about 19% of cases 61). Patients with solid-variant alveolar histology have a lower incidence of PAX-FOXO1 gene fusions than do patients showing classical alveolar histology 62). For the diagnosis of alveolar rhabdomyosarcoma, a FOXO1 gene rearrangement may be detected with good sensitivity and specificity using either fluorescence in situ hybridization or reverse transcription–polymerase chain reaction 63). The alveolar histology that is associated with the PAX7 gene in patients with or without metastatic disease appears to occur at a younger age and may be associated with longer event-free survival rates than those associated with PAX3 gene rearrangements 64). Patients with alveolar histology and the PAX3 gene are older and have a higher incidence of invasive tumor (T2). Around 22% of cases showing alveolar histology have no detectable PAX gene translocation 65). In addition to FOXO1 rearrangements, alveolar tumors are characterized by a lower mutational burden than are fusion-negative tumors, with fewer genes having recurring mutations 66). BCOR and PIK3CA mutations and amplification of MYCN, MIR17HG, and CDK4 have also been described.
- Spindle cell/sclerosing histology: Spindle cell/sclerosing rhabdomyosarcoma has been proposed as a separate entity in the World Health Organization Classification of Tumors of Soft Tissue and Bone 67). For congenital/infantile spindle cell rhabdomyosarcoma, a study reported that 10 of 11 patients showed recurrent fusion genes. Most of these patients had truncal primary tumors, and no paratesticular tumors were found. Novel VGLL2 rearrangements were observed in seven patients (63%), including the VGLL2-CITED2 fusion in four patients and the VGLL2-NCOA2 fusion in two patients 68). Three patients (27%) harbored different NCOA2 gene fusions, including TEAD1-NCOA2 in two patients and SRF-NCOA2 in one patient. All fusion-positive congenital/infantile spindle cell rhabdomyosarcoma patients with available long-term follow-up were alive and well, and no patients developed distant metastases 69). Further study is needed to better define the prevalence and prognostic significance of these gene rearrangements in young children with spindle cell rhabdomyosarcoma. In older children and adults with spindle cell/sclerosing rhabdomyosarcoma, a specific MYOD1 mutation (p.L122R) has been observed in a large proportion of patients 70). Activating PIK3CA mutations are seen in about one-half of the cases, and 60% of these cases have pure sclerosing morphology 71). The presence of the MYOD1 mutation is associated with an increased risk of local and distant failure 72). In one study that included 15 children with MYOD1-mutant tumors, the most common primary site was the head and neck region 73). These patients had sclerosing spindle or mixed histology, and 10 of 15 patients died of disease despite aggressive multimodal therapy.
These findings highlight the important differences between embryonal and alveolar tumors. Data demonstrate that PAX-FOXO1 fusion–positive alveolar tumors are biologically and clinically different from fusion-negative alveolar tumors and embryonal tumors 74). In a study of Intergroup Rhabdomyosarcoma Study Group patients, which captured an entire cohort from a single prospective clinical trial, the outcome for patients with translocation-negative alveolar rhabdomyosarcoma was better than that observed for translocation-positive patients. The outcome was similar to that seen in patients with embryonal rhabdomyosarcoma and demonstrated that fusion status is a critical factor for risk stratification in pediatric rhabdomyosarcoma.
Genome-wide methylation assays can accurately identify PAX3 and PAX7 fusion–positive rhabdomyosarcomas, as well as wild-type and RAS mutant fusion–negative tumors 75).
Response to therapy
It is unlikely that response to induction chemotherapy, as judged by anatomic imaging, correlates with the likelihood of survival in patients with rhabdomyosarcoma, on the basis of the IRSG, Children’s Oncology Group, and International Society of Pediatric Oncology (SIOP) studies that found no association 76). However, an Italian study did find that patient response correlated with likelihood of survival 77). In patients with embryonal rhabdomyosarcoma who had metastases only in the lungs, the Cooperative Weichteilsarkom Studiengruppe assessed the relationship between complete response of the lung metastases at weeks 7 to 10 after chemotherapy and outcome in 53 patients 78). Five-year survival was 68% for 26 complete responders at weeks 7 to 10 versus 36% for 27 patients who achieved complete responses at later time points.
Other studies have investigated response to induction therapy, showing benefit to response. These data are somewhat flawed because therapy is usually tailored on the basis of response and thus, the situation is not as clear as the Children’s Oncology Group data suggests 79).
Response as judged by sequential functional imaging studies with fluorine F 18-fludeoxyglucose positron emission tomography (PET) may be an early indicator of outcome 80) and is under investigation by several pediatric cooperative groups. A retrospective analysis of 107 patients from a single institution examined PET scans performed at baseline, after induction chemotherapy, and after local therapy 81). Standardized uptake value measured at baseline predicted progression-free survival and overall survival, but not local control. A negative scan after induction chemotherapy correlated with statistically significantly better progression-free survival. A positive scan after local therapy predicted worse progression-free survival, overall survival, and local control. PET scans have been shown to be useful in understanding patterns of spread, particularly in patients with extremity disease 82).
Rhabdomyosarcoma complications
Rhabdomyosarcoma can spread from where it started to other areas, making treatment and recovery more difficult.
As with other types of serious cancer, aggressive chemotherapy and radiation for rhabdomyosarcoma can cause substantial side effects, both in the short and long term. Your health care team takes steps to treat and manage these effects as best as possible. And it’s important for you to learn what to watch for and contact your team with any concerns.
Rhabdomyosarcoma causes
Scientists do not know what causes most cases of rhabdomyosarcoma, but they are learning how normal cells become cancerous because of certain changes in their DNA. DNA is the chemical in each of our cells that makes up our genes – the instructions for how our cells function. It is packaged in chromosomes (long strands of DNA in each cell). You normally have 23 pairs of chromosomes in each cell (one set of chromosomes comes from each parent). You usually look like your parents because they are the source of your DNA. But DNA affects more than how you look.
Some genes control when your cells grow, divide into new cells, and die. Genes that help cells grow, divide, or stay alive are called oncogenes. Others that slow down cell division or make cells die at the right time are called tumor suppressor genes. Cancers can be caused by DNA changes that turn on oncogenes or turn off tumor suppressor genes.
Certain genes in a cell can be turned on when bits of DNA are switched from one chromosome to another. This type of change, called a translocation, can happen when a cell is dividing into 2 new cells. This seems to be the cause of most cases of alveolar rhabdomyosarcoma. In these cancers, a small piece of chromosome 2 (or, less often, chromosome 1) ends up on chromosome 13. This moves a gene called PAX3 (or PAX7 if it’s chromosome 1) right next to a gene called FOXO1. The PAX genes play an important role in cell growth while an embryo’s muscle tissue is being formed, but these genes usually shut down once they’re no longer needed. The normal function of the FOXO1 gene is to activate other genes. Moving them together probably activates the PAX genes, which may be what leads to the tumor forming.
Research suggests that embryonal rhabdomyosarcoma develops in a different way. Cells of this tumor have lost a small piece of chromosome 11 that came from the mother, and it has been replaced by a second copy of that part of the chromosome from the father. This seems to make the IGF2 gene on chromosome 11 overactive. The IGF2 gene codes for a protein that can make these tumor cells grow. Other gene changes are probably important in these tumors as well.
Changes in several different genes are usually needed for normal cells to become cancer cells. Scientists have found some other gene changes that set some rhabdomyosarcoma cells apart from normal cells, but there are likely still others that haven’t been found yet.
Scientists now understand many of the gene changes that can lead to rhabdomyosarcoma, but it’s still not clear what causes these changes. Some gene changes can be inherited. Others might just be a random event that sometimes happens inside a cell, without having an outside cause. There are no known lifestyle-related or environmental causes of rhabdomyosarcoma, so it’s important to know that there is nothing children with rhabdomyosarcoma or their parents could have done to prevent these cancers.
Risk factors for rhabdomyosarcoma
A risk factor is anything that affects the chance of having a disease such as cancer. Different cancers have different risk factors.
Most cases of rhabdomyosarcoma occur sporadically, with no recognized predisposing risk factor, with the exception of the following 83):
- Genetic factors:
- Li-Fraumeni cancer susceptibility syndrome (with germline TP53 mutations) 84).
- DICER1 syndrome 85).
- Neurofibromatosis type I 86).
- Costello syndrome (with germline HRAS mutations) 87).
- Beckwith-Wiedemann syndrome (more commonly associated with Wilms tumor and hepatoblastoma) 88).
- Noonan syndrome 89).
- High birth weight and large size for gestational age are associated with an increased incidence of embryonal rhabdomyosarcoma 90).
Lifestyle-related risk factors such as body weight, physical activity, diet, and tobacco use play a major role in many adult cancers. But these factors usually take many years to influence cancer risk, and they are not thought to play much of a role in childhood cancers, including rhabdomyosarcoma.
In most cases, children with rhabdomyosarcoma have no family history of cancer. More research is needed, but the risk of the embryonal type of rhabdomyosarcoma appears to increase in people with a first-degree relative — parent, sibling or child — with cancer, especially when relatives were diagnosed with cancer before the age of 30.
In rare cases, rhabdomyosarcoma may be linked with neurofibromatosis, a genetic disorder that causes tumors to form on nerve tissue. Though more confirming research is needed, in rare cases, rhabdomyosarcoma may be linked with certain inherited syndromes such as Li-Fraumeni syndrome, Beckwith-Wiedemann syndrome or Costello syndrome.
Age and gender
Rhabdomyosarcoma is most common in children younger than 10, but it can also develop in teens and adults. It is slightly more common in boys than in girls.
Inherited conditions
Some people have a tendency to develop certain types of cancer because they have inherited changes in their DNA from their parents. Some rare inherited conditions increase the risk of rhabdomyosarcoma (and usually some other tumors as well).
- Members of families with Li-Fraumeni syndrome are more likely to develop sarcomas (including rhabdomyosarcoma), breast cancer, leukemia, and some other cancers.
- Children with Beckwith-Wiedemann syndrome have a high risk of developing Wilms tumor, a type of kidney cancer, but they are also more likely to develop rhabdomyosarcoma and some other types of childhood cancer.
- Neurofibromatosis type 1, also known as von Recklinghausen disease, usually causes multiple nerve tumors (especially in nerves of the skin), but it also increases the risk of rhabdomyosarcoma.
- Costello syndrome is very rare. Children with this syndrome have high birth weights but then fail to grow well and are short. They also tend to have a large head. They are prone to develop rhabdomyosarcoma as well as some other tumors.
- Noonan syndrome is a condition in which children tend to be short, have heart defects, and can be slower than typical children in developing physical skills and learning things. They are also at higher risk for rhabdomyosarcoma.
These conditions are rare and account for only a small fraction of rhabdomyosarcoma cases. But they suggest that the key to understanding rhabdomyosarcoma might come from studying genes and how they work in very early life to control cell growth and development.
Exposures before birth
Some studies have suggested that being exposed to x-rays before birth might be linked with an increased risk of rhabdomyosarcoma in young children. Parental use of drugs such as marijuana and cocaine has been suggested as a possible risk factor as well. But the studies that have found these links have been small, and more research is needed to see if there is a true link among these factors and rhabdomyosarcoma.
Rhabdomyosarcoma prevention
The risk of many adult cancers can be reduced with certain lifestyle changes (such as staying at a healthy weight or quitting smoking), but at this time there are no known ways to prevent most cancers in children.
The only known risk factors for rhabdomyosarcoma – age, gender, and certain inherited conditions – can’t be changed. There are no proven lifestyle-related or environmental causes of rhabdomyosarcoma, so at this time there is no way to protect against these cancers.
Even though scientists don’t know how to prevent it, most children with rhabdomyosarcoma can be treated successfully.
Rhabdomyosarcoma symptoms
Rhabdomyosarcoma can start nearly anywhere in the body, so there are no symptoms that show up in all cases. The symptoms of rhabdomyosarcoma depend on where the tumor is, how large it is, and if it has spread to other parts of the body.
- When the tumor is in the neck, chest, back, limbs, or groin (including the testicles), the first sign might be a lump or swelling. Sometimes it can cause pain, redness, or other problems.
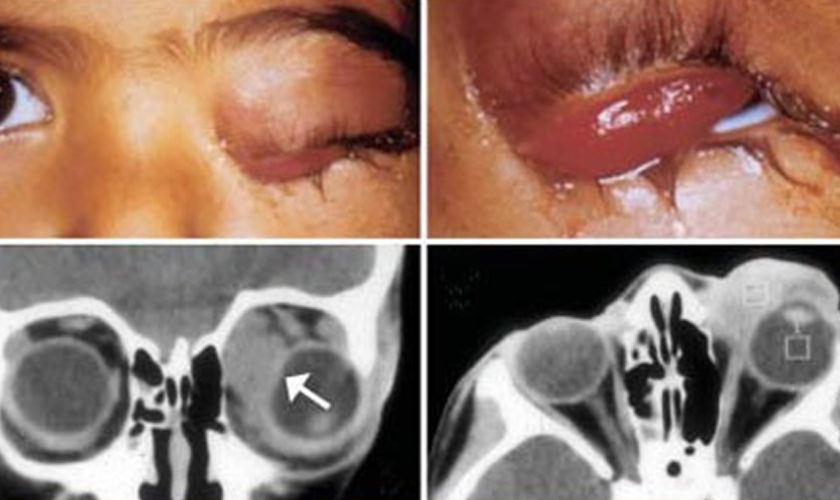
- Tumors around the eye can cause the eye to bulge out or the child to appear to be cross-eyed. Vision might be affected as well.
- Tumors in the ear or nasal sinuses can cause an earache, headache, or sinus congestion.
- Tumors in the bladder or prostate can lead to blood in the urine, while a tumor in the vagina can cause vaginal bleeding. These tumors might grow big enough to make it hard or painful to urinate or have bowel movements.
- Tumors in the abdomen or pelvis can cause vomiting, abdominal pain, or constipation.
rhabdomyosarcoma rarely develops in the bile ducts (small tubes leading from the liver to the intestines), but when it does it can cause yellowing of the eyes or skin (jaundice). - If rhabdomyosarcoma becomes more advanced, it can cause symptoms such as lumps under the skin (often in the neck, under the arm, or in the groin), bone pain, constant cough, weakness, or weight loss.
One or more of these symptoms usually leads parents to bring a child to the doctor. Many of these signs and symptoms are more likely to be caused by something other than rhabdomyosarcoma. For example, children and teens can have bumps or pain from play or sports injuries. Still, if your child has any of these symptoms and they don’t go away within a week or so, check with your doctor so that the cause can be found and treated, if needed.
Can rhabdomyosarcoma be found early?
At this time, there are no widely recommended screening tests for rhabdomyosarcoma. Screening is testing for a disease such as cancer in people who don’t have any symptoms.
Still, rhabdomyosarcoma often causes symptoms that allow it to be found before it has spread to other parts of the body. For example, small tumors that start in the muscles behind the eye often make the eye bulge. Tumors in the nasal cavity often cause nasal congestion, nosebleeds, or bloody mucus. When small lumps form near the surface of the body, children or their parents often see or feel them.
Many cases of rhabdomyosarcoma start in the bladder or other parts of the urinary tract and can cause trouble emptying the bladder or blood in the urine or in diapers. Tumors starting around the testicles in young boys can cause painless swelling that is often noticed early by a parent. In girls with rhabdomyosarcoma of the vagina, the tumor might cause bleeding or a mucus-like discharge from the vagina.
It can be harder to recognize tumors in the arms, legs, and trunks of older children because they often have pain or bumps from sports or play injuries.
There are many other causes of the symptoms above, and most of them are not serious, but it is important to have them checked by a doctor. This includes having your child’s doctor check out any pain, swelling, or lumps that grow quickly or don’t go away after a week or so.
About 1 in 3 of these cancers is found early enough so that all of the visible cancer can be removed completely by surgery. But even when this happens, very small tumors (which cannot be seen, felt, or detected by imaging tests) could already have spread to other parts of the body, which is why other treatments are needed as well.
Families known to carry inherited conditions that raise the risk of rhabdomyosarcoma or that have several family members with cancer (particularly childhood cancers) should talk with their doctors about the possible need for more frequent checkups. It is not common for rhabdomyosarcoma to run in families, but close attention to possible early signs of cancer might help find it early, when treatment is most likely to be successful.
Rhabdomyosarcoma diagnosis
Certain signs and symptoms might suggest that a person has rhabdomyosarcoma, but tests are needed to find out for sure. After the patient is diagnosed with rhabdomyosarcoma, an extensive evaluation to determine the extent of the disease should be performed before instituting therapy. This evaluation typically includes the following:
- Chest x-ray.
- Computed tomography (CT) scan of the chest.
- The European Pediatric Soft Tissue Sarcoma Study Group reviewed 367 patients enrolled in the CCLG-EPSSG-RMS-2005 (NCT00379457) study 91). By prospective study design, patients with indeterminate pulmonary nodules identified on baseline CT scan of the chest (defined as ≤4 pulmonary nodules measuring <5 mm; or 1 nodule measuring ≥5 mm and <10 mm) received the same treatment as did patients with no pulmonary nodules identified on baseline CT of the chest. Rates of event-free survival and overall survival for both groups were the same. The authors concluded that indeterminate pulmonary nodules at diagnosis, as defined in this summary, do not affect outcome in patients with localized rhabdomyosarcoma.
- CT scan of the abdomen and pelvis (for lower extremity or genitourinary primary tumors).
- Magnetic resonance imaging (MRI) of the base of the skull and brain (for parameningeal primary tumors) and of the primary site of other nonparameningeal primary tumors, as appropriate.
- Regional lymph node evaluation.
- CT or MRI: Cross-sectional imaging (CT or MRI scan) of regional lymph nodes should be obtained.
- Lymph node evaluation: Clearly enlarged lymph nodes should be biopsied when possible. Sentinel lymph node biopsy is more accurate than random lymph node sampling and is preferred in patients with extremity and trunk rhabdomyosarcoma, in which enlarged lymph nodes are not revealed on imaging or by physical examination 92). Many studies have demonstrated that sentinel lymph node biopsies can be safely performed in children with rhabdomyosarcoma, and tumor-positive biopsies alter the treatment plan 93). Pathologic evaluation of normal-appearing regional nodes is currently required for all Soft Tissue Sarcoma Committee of the Children’s Oncology Group (COG-STS) study participants with extremity and trunk primary rhabdomyosarcoma. In boys aged 10 years and older with paratesticular rhabdomyosarcoma, retroperitoneal node dissection (ipsilateral nerve sparing) is currently required for normal-appearing lymph nodes, because microscopic tumor is often documented even when the nodes are not enlarged 94). The International Society of Paediatric Oncology Malignant Mesenchymal Tumour Group has confirmed this is a necessary approach 95).
- Positron emission tomography (PET): PET with fluorine F 18-fludeoxyglucose scans can identify areas of possible metastatic disease not seen by other imaging modalities 96). The efficacy of these imaging studies for identifying involved lymph nodes or other sites of disease is important for staging, and PET imaging is recommended on current Soft Tissue Sarcoma Committee of the Children’s Oncology Group treatment protocols.
- Bilateral bone marrow aspirates and biopsies for selected patients.
- Bone scan for selected patients.
A retrospective study of 1,687 children with rhabdomyosarcoma enrolled in Intergroup Rhabdomyosarcoma Study Group (IRSG) and COG studies from 1991 to 2004 suggests those with localized negative regional lymph nodes, noninvasive embryonal tumors, and Group I alveolar tumors (about one-third of patients) can have limited staging procedures that eliminate bone marrow and bone scan examinations at diagnosis 97).
Medical history and physical exam
If your child has symptoms that could be from rhabdomyosarcoma (or another type of tumor), the doctor will want to get a complete medical history to find out more about the symptoms and how long your child has had them. The doctor will also examine your child to look for possible signs of rhabdomyosarcoma or other health problems. For example, the doctor might be able to see or feel an abnormal lump or swelling.
If the doctor suspects your child might have rhabdomyosarcoma (or another type of tumor), tests will be needed to find out. These might include imaging tests, biopsies, and/or lab tests.
Imaging tests
Imaging tests use x-rays, magnetic fields, radioactive substances, or sound waves to create pictures of the inside of the body. Imaging tests can be done for a number of reasons, including:
- To help find out if a suspicious area might be cancer
- To determine the extent of a tumor or learn how far a cancer has spread
- To help determine if treatment is working
People who have or may have rhabdomyosarcoma will get one or more of these tests.
Plain x-rays
X-rays are sometimes used to look for tumors, but their use is limited mainly to looking at bones because they don’t show much detail in internal organs. A chest x-ray is sometimes done to look for cancer that might have spread to the lungs, although it isn’t needed if a chest CT scan is being done.
Computed tomography (CT) scan
The CT scan uses x-rays to make detailed cross-sectional images of parts of the body, including soft tissues such as muscles. Instead of taking one picture, like a regular x-ray, a CT scanner takes many pictures as it rotates around your child while he or she lies on a table. A computer then combines these pictures into images of slices of the part of the body being studied.
This test can often show a tumor in detail, including how large it is and if it has grown into nearby structures. It can also be used to look at nearby lymph nodes, as well as the lungs or other areas of the body where the cancer might have spread.
Before the scan, your child may be asked to drink a contrast solution and/or get an intravenous (IV) injection of a contrast dye that will help better outline abnormal areas. Your child may need an IV line for the contrast dye. The dye can cause some flushing (a feeling of warmth, especially in the face). Some people are allergic and get hives. Rarely, more serious reactions like trouble breathing or low blood pressure can occur. Be sure to tell the doctor if your child has any allergies (especially to iodine or shellfish) or has ever had a reaction to any contrast material used for x-rays.
CT scans take longer than regular x-rays. A CT scanner has been described as a large donut, with a narrow table that slides in and out of the middle opening. Your child will need to lie still on the table while the scan is being done. Younger children may be given medicine to help keep them calm or even asleep during the test.
Magnetic resonance imaging (MRI) scan
Like CT scans, MRI scans give detailed images of soft tissues in the body. But MRI scans use radio waves and strong magnets to create the images instead of x-rays. A contrast material called gadolinium may be injected into a vein before the scan to help show details better. This contrast material usually does not cause allergic reactions.
This test might be used instead of a CT scan to look at the tumor and the tissues around it. MRI is especially useful if the tumor is in certain parts of the body, such as the head and neck, an arm or leg, or the pelvis. MRI scans can help determine the exact extent of a tumor, because they can show the muscle, fat, and connective tissue around the tumor in great detail. This is important when planning surgery or radiation therapy. MRI is also very useful if your child’s doctor is concerned about possible spread to the spinal cord or brain.
MRI scans take longer than CT scans – often up to an hour. Your child may have to lie on a table that slides inside a narrow tube, which is confining and can be distressing. The test also requires a person to stay still for several minutes at a time. Newer, more open MRI machines, which are less confining, might be an option, but the test still requires staying still for long periods of time. The MRI machine also makes loud buzzing and clicking noises that can be disturbing. Sometimes, younger children are given medicine to help keep them calm or even asleep during the test.
Bone scan
A bone scan can help show if a cancer has spread to the bones, and is often part of the workup for anyone with rhabdomyosarcoma. This test is useful because it provides a picture of the entire skeleton at once.
For this test, a small amount of low-level radioactive material is injected into a vein (IV). The amount of radioactivity used is very low and will pass out of the body within a day or so. Over a couple of hours, the substance settles in abnormal areas of bone throughout the body. Your child then lies on a table for about 30 minutes while a special camera detects the radioactivity and creates a picture of the skeleton. Younger children can be given medicine to help keep them calm or even asleep during the test.
Areas of active bone changes attract the radioactivity and show up as “hot spots” on the scan. These areas may suggest cancer in an area, but other bone diseases can also cause the same pattern, so other tests such as plain x-rays or MRI scans, or even a bone biopsy might be needed.
Positron emission tomography (PET) scan
For a PET scan, a radioactive substance (usually a type of sugar related to glucose, known as FDG) is injected into the blood. The amount of radioactivity used is very low and will pass out of the body in a day or so. Because cancer cells in the body are growing quickly, they will absorb large amounts of the sugar.
After about an hour, your child will lie on a table in the PET scanner for about 30 minutes while a special camera creates a picture of areas of radioactivity in the body. The picture is not detailed like a CT or MRI scan, but it provides helpful information about the whole body.
PET scans are not used routinely to help diagnose rhabdomyosarcoma, but they can sometimes be helpful in finding out if suspicious areas seen on other imaging tests (such as bone scans or CT scans) are tumors. PET scans can also be repeated during treatment to monitor the cancer over time.
Some machines can do a PET and CT scan at the same time (PET/CT scan). This lets the doctor compare areas of higher radioactivity on the PET scan with the more detailed appearance of that area on the CT scan.
Ultrasound
Ultrasound uses sound waves and their echoes to make a picture of internal organs or tumors. For this test, a small, microphone-like instrument called a transducer is moved around on the skin (which is first lubricated with gel). It gives off sound waves and picks up the echoes as they bounce off the organs. The echoes are converted by a computer into an image on a screen.
Ultrasound can be used to see if tumors in the pelvis (such as prostate or bladder tumors) are growing or shrinking over time. (This test can’t be used to look at tumors in the chest because the ribs block the sound waves.)
This is an easy test to have, and it uses no radiation. Your child simply lies on a table, and a doctor or technician moves the transducer over the part of the body being looked at.
Biopsy
The results of imaging tests might strongly suggest that someone has rhabdomyosarcoma, but a biopsy (removing some of the tumor for viewing under a microscope and other lab testing) is the only way to be certain. Usually several different kinds of lab tests are done on the biopsy sample to sort out what kind of tumor it is.
Biopsies can be done in several ways. The approach used depends on where the tumor is, the age of the patient, and the expertise and experience of the doctor doing the biopsy.
Surgical biopsy
The most common biopsy approach is to remove a small piece of tumor during surgery while the patient is under general anesthesia (asleep). In some cases, nearby lymph nodes are also removed to see if the tumor has spread to them. The samples are then sent to a lab and tested.
Needle biopsies
If for some reason a surgical biopsy can’t be done, a less invasive biopsy using a thin, hollow needle may be done. There are 2 kinds of needle biopsies, each of which has pros and cons.
- Core needle biopsy: For a core needle biopsy, the doctor inserts a hollow needle into the tumor to withdraw a piece of it (known as a core sample). If the tumor is just under the skin, the doctor can guide the needle into the tumor by touch. But if the tumor is deep inside the body, imaging tests such as ultrasound or CT scans might be needed to help guide the needle into place. The removed core sample is then sent to the lab for testing.
The main advantage of a core needle biopsy is that it does not require surgery, so there is no large incision. Depending on where the tumor is, adults and older children might not need general anesthesia (where they are asleep for the biopsy), but some younger children might. On the other hand, the specimen is smaller than with a surgical biopsy, and if the needle isn’t aimed correctly, it might miss the cancer. If the specimen is not a good sample of the tumor, another biopsy will be needed.
- Fine needle aspiration (FNA) biopsy: For this technique, the doctor uses a very thin, hollow needle attached to a syringe to withdraw (aspirate) a small tumor sample. An FNA biopsy is best suited for tumors that can be reached easily (such as those just under the skin), although it can also be used for tumors deeper in the body.
The downside of FNA is that the sample is very, very small. The pathologist must be experienced with this technique and be able to decide which lab tests will be most helpful on a very small sample. In cancer centers that have the experience to extract the most information from very small amounts of tissue, FNA can be a valuable – though certainly not foolproof – way to diagnose rhabdomyosarcoma, but it is not usually the preferred biopsy technique.
Bone marrow aspiration and biopsy
These tests aren’t used to diagnose rhabdomyosarcoma, but they are often done after the diagnosis to find out if the tumor has spread to the bone marrow (the soft inner parts of certain bones).
The 2 tests are usually done at the same time. The samples are usually taken from the back of both of the pelvic (hip) bones, but in some patients they may be taken from other bones.
These tests might be done during the surgery to treat the main tumor (while the child is still under anesthesia), or they might be done as a separate procedure.
If the bone marrow aspiration is being done as a separate procedure, the child lies on a table (on his or her side or belly). After cleaning the skin over the hip, the doctor numbs the area and the surface of the bone with local anesthetic, which can briefly sting or burn. In most cases, the child is also given other medicines to help them relax or even be asleep during the procedure. A thin, hollow needle is then inserted into the bone, and a syringe is used to suck out a small amount of liquid bone marrow.
A bone marrow biopsy is usually done just after the aspiration. Small pieces of bone and marrow are removed with a slightly larger needle that is pushed down into the bone. Once the biopsy is done, pressure will be applied to the site to help stop any bleeding.
The samples of bone and marrow are sent to the lab, where they are looked at and tested for cancer cells.
Lumbar puncture (spinal tap)
Lumbar puncture is not a common test for rhabdomyosarcoma, but it might be done for tumors in the head near the covering of the brain (the meninges). This test is used to look for cancer cells in the cerebrospinal fluid (CSF), which is the liquid that bathes the brain and spinal cord.
For this test, the doctor first numbs an area in the lower part of the back near the spine. The doctor may also recommend that the child be given something to make him or her sleep so the spinal tap can be done without difficulty or causing harm. A small, hollow needle is then inserted between the bones of the spine to withdraw some of the fluid, which is then sent to the lab for testing.
Lab tests on the biopsy samples
A doctor called a pathologist looks at the biopsy samples under a microscope to see if they contain cancer cells. If cancer is found, the next step is to figure out if it is rhabdomyosarcoma. In rare cases, the pathologist can see that the cancer cells have small muscle striations, which confirms that the cancer is rhabdomyosarcoma. But most often, other lab tests are needed to be sure.
The pathologist might use special stains on the samples to identify the type of tumor. The stains contain special proteins (antibodies) that attach to substances in rhabdomyosarcoma cells but not to other cancers. The stains produce a distinct color that can be seen under a microscope. This lets the pathologist know that the tumor is a rhabdomyosarcoma.
Sometimes the tumor will also be tested for gene or chromosome changes.
If a diagnosis of rhabdomyosarcoma is made, the pathologist will also use these tests to help determine which kind of rhabdomyosarcoma it is. This is important because it affects how the cancer is treated. For example, alveolar rhabdomyosarcoma, which tends to be more aggressive, typically requires more intense treatment than embryonal rhabdomyosarcoma.
Blood tests
No blood test can be used to diagnose rhabdomyosarcoma. But certain blood tests may be helpful once a diagnosis has been made.
A complete blood count (CBC) measures the levels of white blood cells, red blood cells, and platelets in the blood. If the CBC result is abnormal at the time of diagnosis it could mean the cancer has spread to the bone marrow, where these blood cells are made.
Standard blood tests are done often to check a child’s general health both before treatment (especially before surgery) and during treatment (such as chemotherapy) to look for possible problems or side effects. These tests often include a CBC to monitor bone marrow function and blood chemistry tests to measure how well the liver and kidneys are working.
Rhabdomyosarcoma Stage
Before a suspected tumor mass is biopsied, imaging studies of the mass and baseline laboratory studies should be obtained. Once rhabdomyosarcoma has been diagnosed and the type of rhabdomyosarcoma identified, doctors need to assess, as accurately as possible, how much cancer there is and where it has spread. The answers to these questions are expressed in a standard kind of shorthand known as staging.
The prognosis (outlook) for people with cancer depends, to a large extent, on the cancer’s stage. The stage of a cancer is one of the most important factors in choosing treatment.
Your child’s doctors will use the results of the imaging tests and biopsies and the direct examination of the organs during surgery to learn how far the cancer has spread. If there is any doubt about the extent of the cancer, more biopsies may be done on tissues at the edge of the tumor, nearby lymph nodes, and any suspicious lumps in other parts of the body.
To stage rhabdomyosarcoma, doctors first determine 3 key pieces of information:
- The type of rhabdomyosarcoma (embryonal or alveolar)
- The TNM stage
- The clinical group
These factors are then used to divide patients into risk groups, which then are used to guide treatment.
Rhabdomyosarcoma is staged differently from most other cancers, and it can be confusing. If you have any questions about the staging or risk groups, ask the doctor or nurse to explain it to you in a way you understand.
The TNM stage
The TNM stage is determined before treatment starts, and is based on 3 key pieces of information:
- T: The characteristics of the main tumor (location and size)
- N: Whether the cancer has spread to nearby lymph nodes (bean-sized collections of immune system cells)
- M: Whether it has metastasized (spread) to distant parts of the body
These factors are combined to determine an overall stage:
Stage 1
The tumor started in a favorable area:
- The orbit (area around the eye)
- The head and neck area, except for parameningeal sites (areas next to the membranes covering the brain, such as the nasal passages and nearby sinuses, middle ear, and the uppermost part of the throat)
- A genital or urinary site, except the bladder or prostate gland
- Bile ducts (tubes leading from the liver to the intestines)
The tumor can be any size. It may have grown into nearby areas and/or spread to nearby lymph nodes, but it has not spread to distant parts of the body.
Stage 2
The tumor started in an unfavorable site:
- The bladder or prostate
- An arm or leg
- A parameningeal site (an area next to the membranes covering the brain, such as the nasal passages and nearby sinuses, middle ear, or the uppermost part of the throat)
- Any other part of the body not mentioned in stage 1
The tumor is 5 cm (about 2 inches) or smaller across and there is no evidence that it has spread to nearby lymph nodes or distant parts of the body.
Stage 3
The tumor started in an unfavorable site:
- The bladder or prostate
- An arm or leg
- A parameningeal site (an area next to the membranes covering the brain, such as the nasal passages and nearby sinuses, middle ear, or the uppermost part of the throat)
- Any other part of the body not mentioned in stage 1
And one of the following applies:
- The tumor is 5 cm across or smaller but has spread to nearby lymph nodes
- The tumor is larger than 5 cm across and may or may not have spread to nearby lymph nodes
In either case, the cancer has not spread to distant parts of the body.
Stage 4
The tumor can have started anywhere in the body and can be of any size. It has spread to distant parts of the body such as the lungs, liver, bones, or bone marrow.
Clinical group staging
The clinical group is based on the extent of the disease and how completely it is removed during initial surgery. The groups are defined as follows.
Group I
This group includes children with localized rhabdomyosarcoma (the cancer has not spread to nearby lymph nodes or to distant sites in the body) that is removed completely by surgery.
About 10% to 15% of rhabdomyosarcoma patients are in group I.
Group II
This group includes children who have had all of the visible cancer removed by surgery, but cancer cells have been found at the edges (margins) of the removed specimen (meaning that there may have been a small amount of cancer left behind), in the nearby lymph nodes, or in both places. In all cases, as much of the cancer has been removed as possible.
About 20% of rhabdomyosarcoma patients are in group II.
Group III
These children have tumors that could not be removed completely. Some tumor was left behind that could be seen with the naked eye. The cancer may have spread to nearby lymph nodes, but there is no sign that it has spread to distant organs.
About 50% of rhabdomyosarcoma patients are in group III.
Group IV
At the time of diagnosis, these children have evidence of distant cancer spread to places such as the lungs, liver, bones, bone marrow, or to distant muscles or lymph nodes.
About 15% to 20% of rhabdomyosarcoma patients are in group IV.
Risk groups
Using the information about the type of rhabdomyosarcoma, the TNM stage, and the clinical group, doctors classify patients into 3 risk groups. Information about risk groups helps doctors decide how aggressive treatment should be.
The risk groups are based on what has been learned from previous research on patients’ outcomes. The groups discussed here are based on the most current information, but these may change in the future as safer and more effective treatments are developed.
Low-risk group
About 1 in 3 children with rhabdomyosarcoma falls into the low-risk group. It includes:
- Children with TNM stage 1 embryonal rhabdomyosarcomas that fall into clinical groups I, II, or III
- Children with stage 2 or 3 embryonal rhabdomyosarcoma who are in clinical groups I or II
Intermediate-risk group
About half of children of rhabdomyosarcoma fall into the intermediate-risk group. It includes:
- Children with stage 2 or 3 embryonal rhabdomyosarcoma who are in clinical group III
- Children with alveolar rhabdomyosarcoma that has not spread to distant parts of the body (stage 1, 2, or 3)
High-risk group
This group includes:
Children with widespread (stage 4) rhabdomyosarcoma (embryonal rhabdomyosarcoma or alveolar rhabdomyosarcoma)
Rhabdomyosarcoma survival rate
Survival rates are often used by doctors as a standard way of discussing a person’s prognosis (outlook). Some people may want to know the survival statistics for those in similar situations, while others may not find the numbers helpful, or may even not want to know them.
When discussing cancer survival statistics, doctors often use a number called the 5-year survival rate. The 5-year survival rate refers to the percentage of patients who live at least 5 years after their cancer is diagnosed. Of course, many people live much longer than 5 years (and many are cured).
To get 5-year survival rates, doctors have to look at people who were treated at least 5 years ago. Improvements in treatment since then might result in a better outlook for patients being diagnosed with rhabdomyosarcoma now.
Survival rates are often based on previous outcomes of large numbers of people who had the disease, but they can’t predict what will happen in any person’s case. For a person with rhabdomyosarcoma, the risk group is important in estimating their outlook. But many other factors can also affect a person’s outlook, such as their age, the location of the tumor, certain gene changes in the cancer cells, and how well the cancer responds to treatment.
Here are general survival statistics based on risk groups. These numbers come from large clinical trials treating children with rhabdomyosarcoma in the 1980s and 1990s.
Low-risk group
Overall, the 5-year survival rate for children in the low-risk group is over 90%. Most of these children will be cured.
Intermediate-risk group
For those in the intermediate-risk group, the 5-year survival rates range from about 60% to about 80%. The rate varies based on tumor location, stage, and the age of the child (children aged 1 to 9 tend to do better than older or younger children).
High-risk group
If the cancer has spread widely, the 5-year survival rate is generally around 20% to 40%. Again, it’s important to note that other factors, such as the patient’s age and the site and type of tumor will affect these numbers. For example, children with embryonal rhabdomyosarcoma and limited spread (to only 1 or 2 distant sites) have a higher 5-year survival rate. Also, children 1 to 9 years of age tend to have a better outlook than younger or older patients.
Even when taking risk groups and other factors into account, survival rates are at best rough estimates. Your child’s doctor is your best source of information on this topic, as he or she knows your situation best.
Rhabdomyosarcoma treatment
Once rhabdomyosarcoma has been found and staged, the cancer care team will talk with you about treatment options. It’s important to be sure you understand your child’s options as well as their possible side effects to help make the decision that’s the best fit for your child. If there is anything you don’t understand, ask to have it explained.
The treatment and prognosis (outlook) for patients with rhabdomyosarcoma depend to a large extent on the type of rhabdomyosarcoma and on how much of it can be removed with surgery. This is why it’s very important for patients to be diagnosed and treated by doctors who have experience with rhabdomyosarcoma. Children with rhabdomyosarcoma are best treated in a cancer center where there is experience and expertise in treating childhood cancers, such as in centers who are members of the Children’s Oncology Group.
For children and teens, a team approach is recommended that includes specialists at a children’s cancer, as well as the child’s pediatrician. For adults with rhabdomyosarcoma, the treatment team typically includes specialists at a major cancer center, as well as the patient’s primary care doctor. Doctors on the treatment team might include:
- An orthopedic surgeon (a surgeon who specializes in muscles and bones) who is experienced in treating rhabdomyosarcoma
- A medical or pediatric oncologist (a doctor who treats cancer with chemotherapy and other drugs)
- A radiation oncologist (a doctor who treats cancer with radiation therapy)
- A pathologist (a doctor specializing in using lab tests to diagnose and classify diseases)
- A physiatrist (a doctor who directs a person’s rehabilitation and physical therapy after treatment)
The team might also include other doctors, as well as physician assistants, nurse practitioners, nurses, psychologists, social workers, physical therapists and other rehabilitation specialists, and other health professionals. Going through cancer treatment often means meeting lots of specialists and learning about parts of the medical system you probably haven’t been exposed to before.
The types of treatment that can be used for rhabdomyosarcoma include:
- Surgery
- Chemotherapy
- Radiation therapy
- High-dose chemotherapy and stem cell transplant (very rarely)
- Clinical trials. Clinical trials are studies to investigate new ways of treating cancer. Many of the advances in treating pediatric cancers, including rhabdomyosarcoma, come from clinical trials of the Children’s Oncology Group, which has more than 200 participating medical institutions from the United States and other countries.
All children and adults with rhabdomyosarcoma will be treated with surgery to remove the tumor if it can be done without causing major damage or disfigurement. If this isn’t possible, chemotherapy and/or radiation therapy may be used first to try to shrink the tumor. If it shrinks enough, surgery can be done at this point. The goal of surgery is to remove the tumor completely, but this isn’t always possible.
Whether the tumor appears to have been removed completely or not, all patients with rhabdomyosarcoma need chemotherapy. Without it, it’s very likely that the cancer will come back in distant parts in the body because small amounts of cancer have almost always reached other parts of the body when the cancer is first found.
If cancer is left behind after surgery or if the cancer has some less favorable traits and it hasn’t spread to distant sites (as is the case most of the time), radiation therapy will also be given.
Many of these treatments can be used again if the cancer continues to grow or if it comes back later on.
All of these treatments can have side effects, but many of them can be made less troublesome. Your medical team will help you take care of the side effects and help you understand and deal with the medical problems, stress, and other issues related to treatment.
Because many of these things can be more complex for cancer in children, many people will be involved in your child’s overall care. As a parent, taking care of a child with cancer can be a very big job. It’s important to remember that you will have a lot of help. It’s also important for you to know that the health care professionals who treat children with rhabdomyosarcoma are using the experience and knowledge gained from many decades of detailed scientific study of treating this disease.
Embryonal rhabdomyosarcoma treatment
This type tends to occur in children under 15 and in the head and neck region and the bladder or genital area.
With embryonal rhabdomyosarcoma, the position in the body can decide the treatment.
Surgery may be the initial treatment. Chemotherapy tends to work well for this type of sarcoma. You may have chemotherapy before surgery to shrink a tumor and make it easier to remove. Or it may be given after surgery to try to reduce the chance of the tumor coming back.
If the tumor is in a place where it is not possible to remove it completely with surgery (for example, behind the nose or in the eye socket), your doctor will treat it with a combination of radiotherapy and chemotherapy.
Alveolar rhabdomyosarcoma treatment
This occurs in the arms or legs of older children and young people but can also occur in the muscles of the trunk.
With alveolar rhabdomyosarcoma, usually the tumor is removed with surgery and then you have radiotherapy to the area where the tumor was.
The radiotherapy aims to reduce the chance of the tumor coming back in the same place. Chemotherapy is usually be given before or after the surgery.
Anaplastic rhabdomyosarcoma and undifferentiated sarcoma treatment
This type of rhabdomyosarcoma usually occurs in adults and in the arms or legs.
With anaplastic rhabdomyosarcoma and undifferentiated sarcoma, usually the treatment is surgery and then radiotherapy.
The radiotherapy aims to reduce the chance of the tumor coming back in the same place. Chemotherapy tends not to work very well and so is not usually used.
Coping and support
A diagnosis of rhabdomyosarcoma can be frightening — especially for the family of a newly diagnosed child. Coping with a diagnosis of a rare cancer can be especially difficult, both practically and emotionally. Being well informed about your cancer and its treatment can make it easier to make decisions and cope with what happens. With time you’ll find ways to cope with the distress and uncertainty of cancer. Until then, you may find it helps to:
- Learn enough about rhabdomyosarcoma to make decisions about care. Ask your doctor about this sarcoma, including treatment options. As you learn more, you may become more confident in understanding and making decisions about treatment options. If your child has cancer, ask the health care team for guidance on sharing this information in a caring and age-appropriate way.
- Keep friends and family close. Keeping your close relationships strong can help you deal with cancer. Friends and relatives can provide the practical support you’ll need, such as helping take care of your house if your child is in hospital. And they can serve as emotional support when you feel overwhelmed.
- Ask about mental health support. The concern and understanding of a counselor, medical social worker, psychologist or other mental health professional also may help you. If your child has cancer, ask your health care team for advice on providing emotional and social support and options for professional mental health support. You can also check online for a cancer organization, such as the National Cancer Institute 98) or the American Cancer Society 99), that lists support services.
References [ + ]

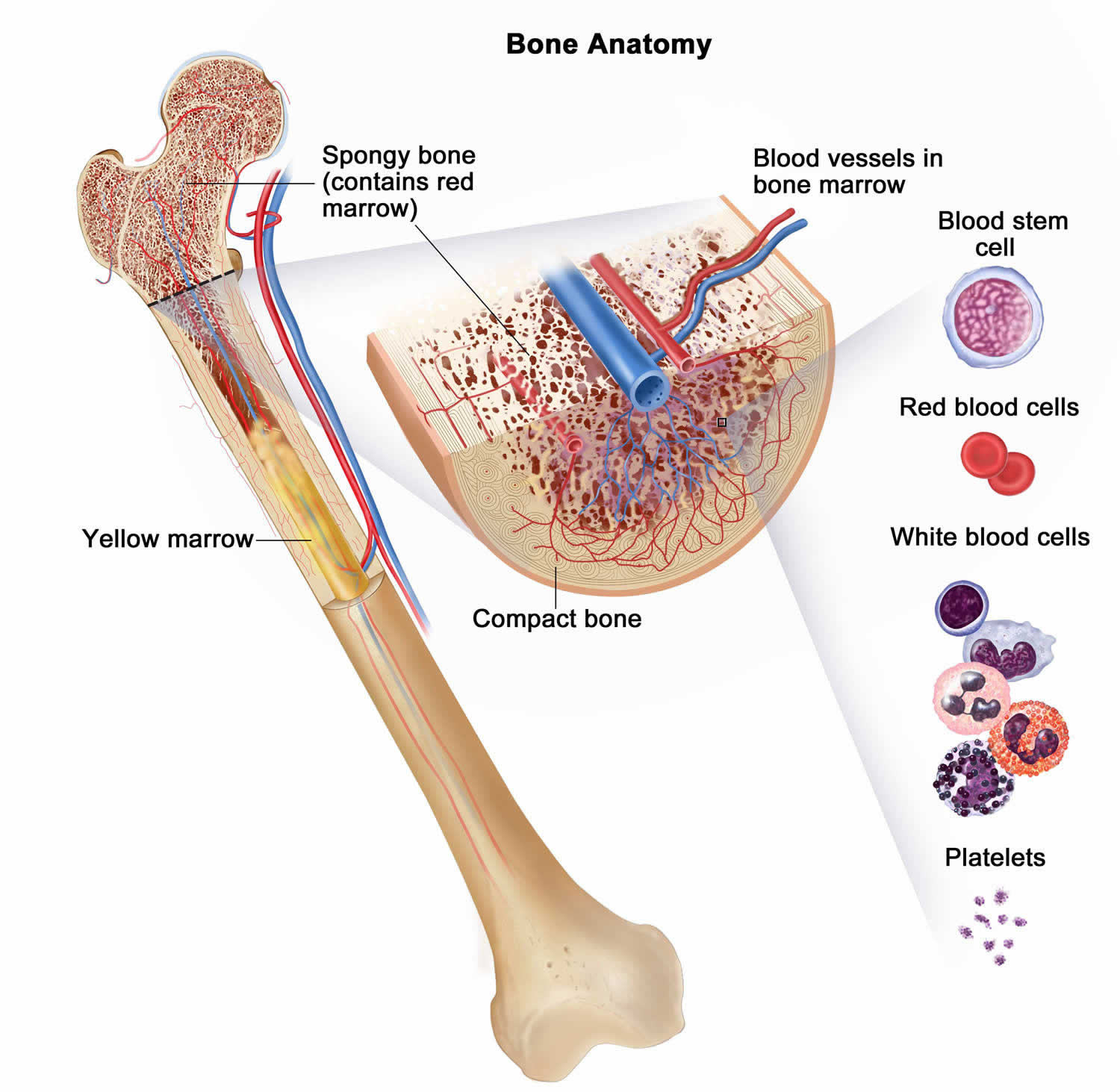
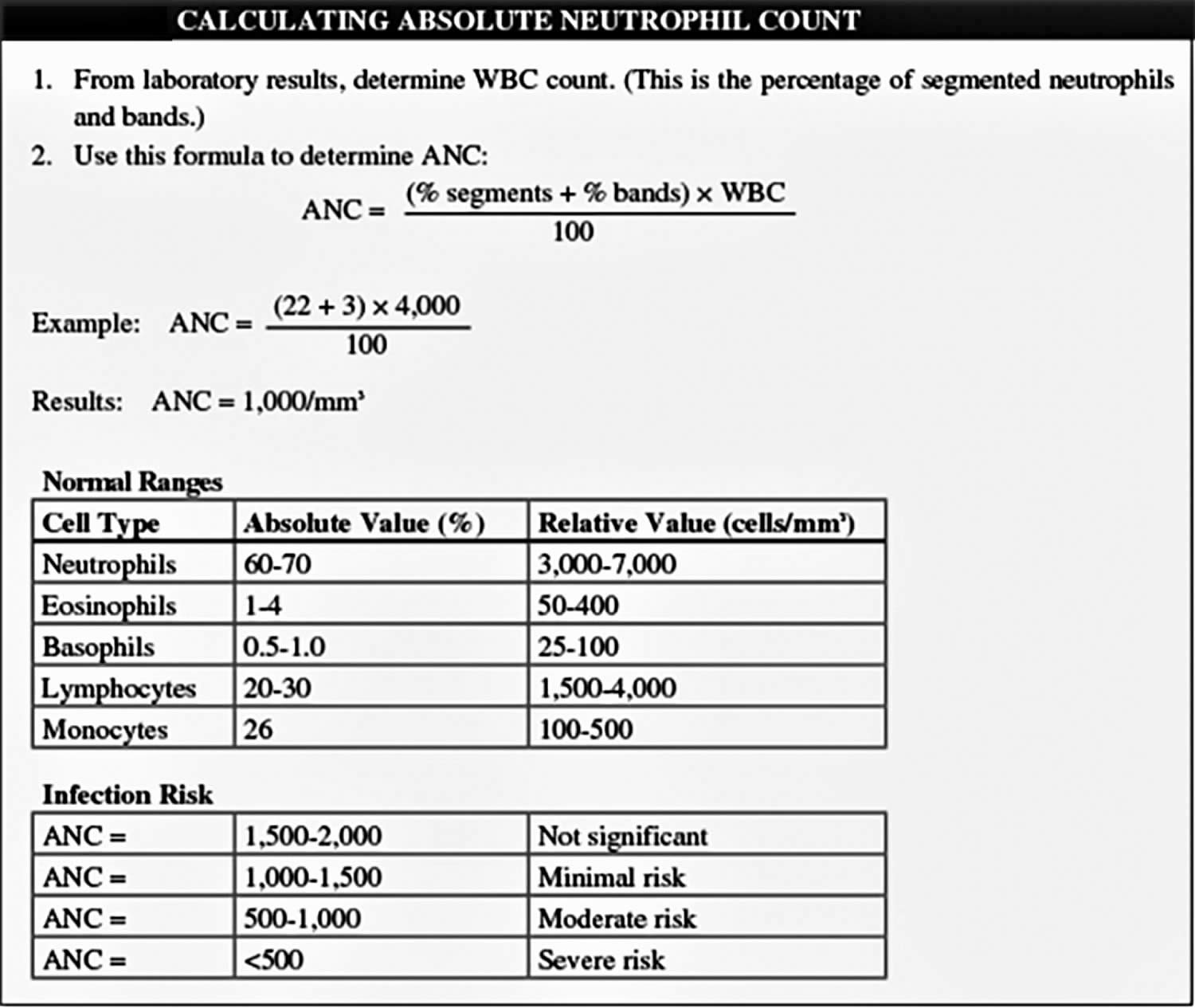

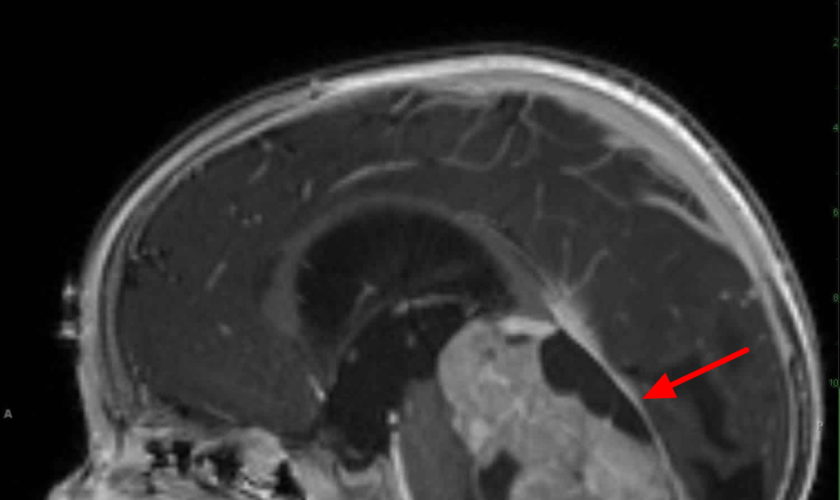
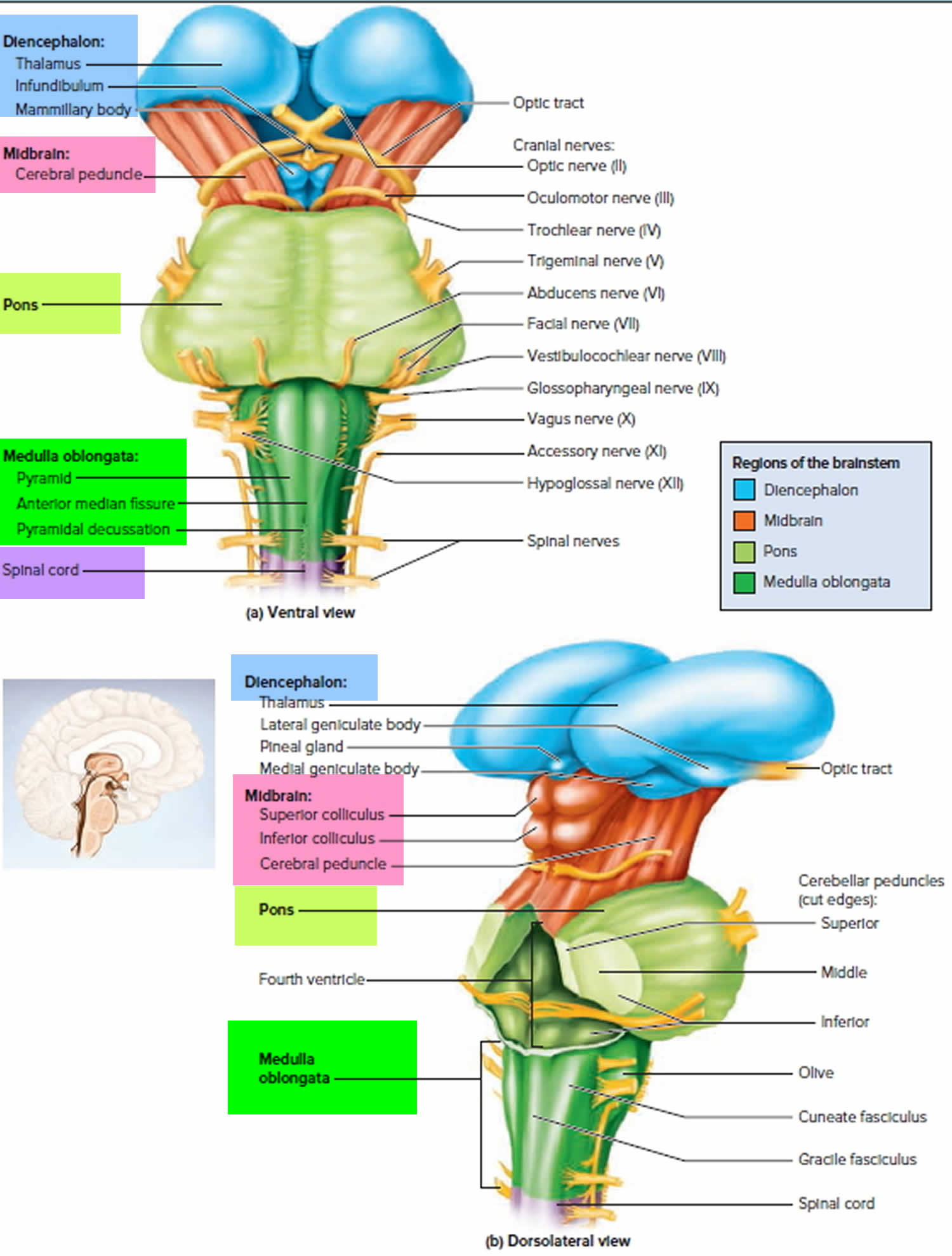
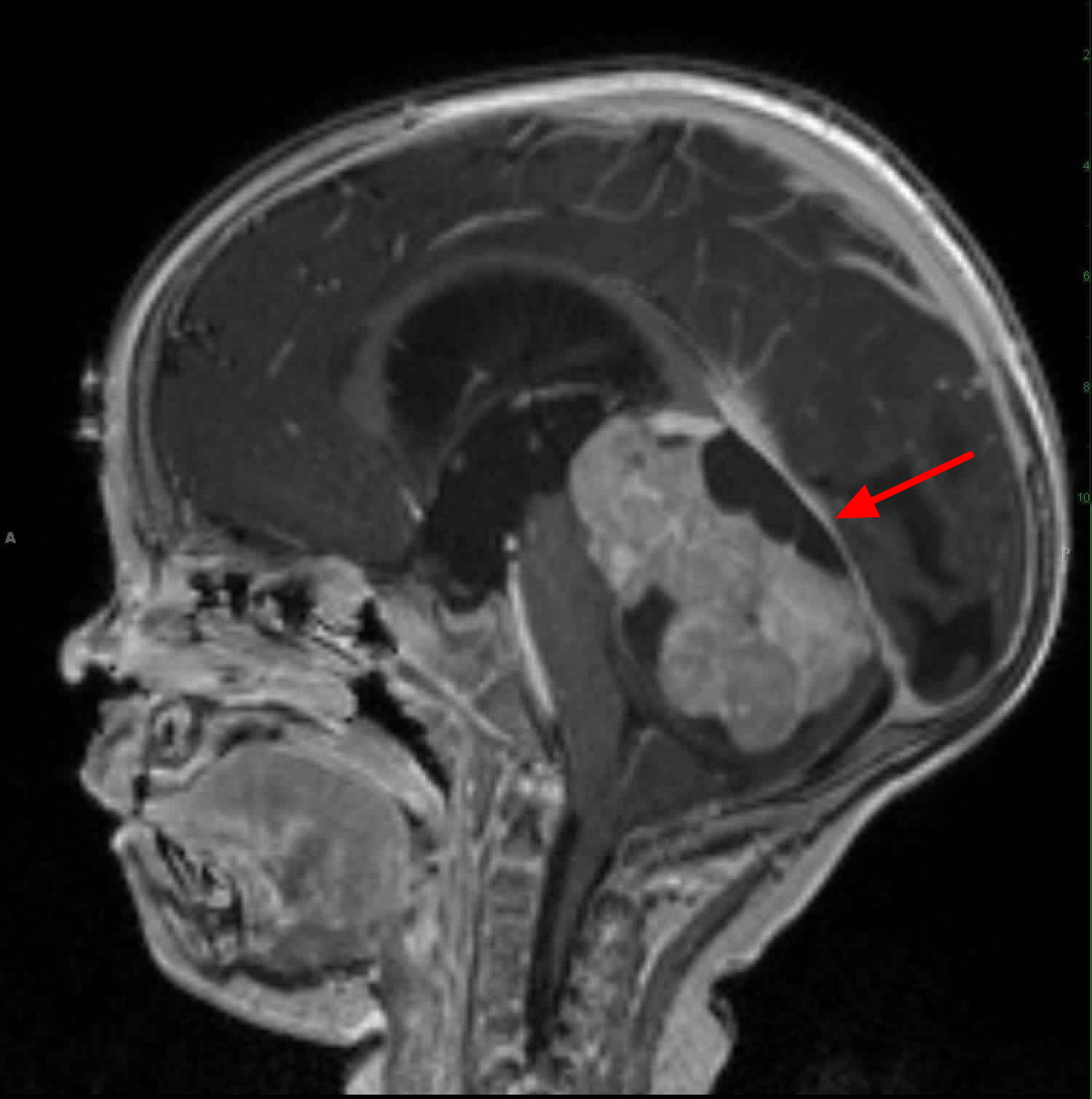


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}