




Delayed puberty
Delayed puberty is when the physical signs of puberty do not appear by age 13 for girls and age 14 for boys. Puberty is considered delayed when there are no signs of breast development by 13 years of age in girls or testicular enlargement by 14 years of age in boys 1). Puberty is the time when a child’s body starts to change to an adult’s. Normally, these changes begin in girls when they’re between 8 and 14 years old. In boys, they start between the ages of 9 and 15. This wide range in age is normal, and it’s why kids may develop several years earlier or later than many of their friends. Sometimes, though, kids pass this normal age range for puberty without showing any signs of body changes. This is called delayed puberty. Clinicians should suspect pubertal delay if there is halting or regression of pubertal development. In girls with initial pubertal changes, absence of menarche (a girl’s first period) by 15 years of age is also concerning 2).
Signs of delayed puberty in boys include:
- the penis and testicles not starting to grow larger by age 14
- genital growth that takes longer than 5 years
- short stature compared with their peers, who now are growing faster
In girls, signs include:
- no breast development by age 14
- not starting to menstruate within 5 years of when breasts start to grow or by age 16
Delayed puberty, which is more common in boys, can happen for different reasons. Many children with delayed puberty will eventually go through an otherwise normal puberty, just at a late age. Delayed puberty can be hereditary with a condition called constitutional delay of puberty; meaning that the late onset of puberty may run in families. However, delayed puberty may also be due to chromosomal abnormalities, genetic disorders, chronic illnesses, or tumors that damage the pituitary gland or the hypothalamus, which affect maturation.
Delayed puberty can cause significant psychological distress and low self-esteem 3). Doctors usually can help teens with delayed puberty develop so they can catch up with their peers.
Delayed puberty treatment depends on the cause. Children with constitutional delay of growth and puberty don’t need treatment. They will eventually catch up to other children their age.
Medicine might be needed if puberty is very delayed or there is a hormone problem. If your child has a health problem or is underweight, treating the condition that is causing delayed puberty can help.
Girls should see the doctor if they have:
- Signs of puberty before eight years of age
- No signs of puberty by 13 years of age
- No menstrual period by 15 years of age
- Puberty that starts but then stops
Boys should see the doctor if they have:
- Signs of puberty before nine years of age
- No signs of puberty by 14 years of age
- Puberty that starts but then stops
What is normal puberty?
Puberty is when a child starts to mature into an adult. Hormones (estrogen and testosterone) cause the body to change during puberty. Normal puberty starts in the brain. The brain tells the ovaries (in girls) and testicles (in boys) to make estrogen and testosterone. The adrenal glands sit on top of the kidneys. They make testosterone-like hormones.
- For girls, breasts normally start to grow between eight and 13 years of age. Body odor, a growth spurt, pubic and underarm hair are other signs of puberty in girls. A girl’s first menstrual period usually happens about 2.5 years after the breasts start to grow. On average, girls have their first period at 12 or 13 years of age.
- For boys, the testicles start to grow between nine and 14 years of age. Body odor, a growth spurt, pubic and underarm hair, voice changes, and facial hair follow.
The physical changes of puberty are a result of gonadal sex hormone production, the start of which (gonadarche) indicates pubertal onset. Gonadarche is triggered by the pulsatile release of gonadotropin-releasing hormone, which activates the hypothalamic-pituitary-gonadal axis 4). Adrenarche (i.e., adrenal androgen production leading to pubic and axillary hair, body odor, and mild acne) is a separate but usually concurrent process and does not in itself indicate true pubertal onset in boys or girls 5).
In girls, signs of puberty include:
- breast development
- pubic or underarm hair development
- rapid height growth (a growth spurt)
- wider hips and a curvier body shape
- start of menstruation (periods)
In boys, signs include:
- enlargement of the testicles and penis
- pubic, underarm, or facial hair development
- rapid height growth (a growth spurt)
- broader shoulders and a more muscular build
- voice deepening
These changes are caused by the sex hormones testosterone (in boys) and estrogen (in girls) that their bodies start making in much larger amounts than before.
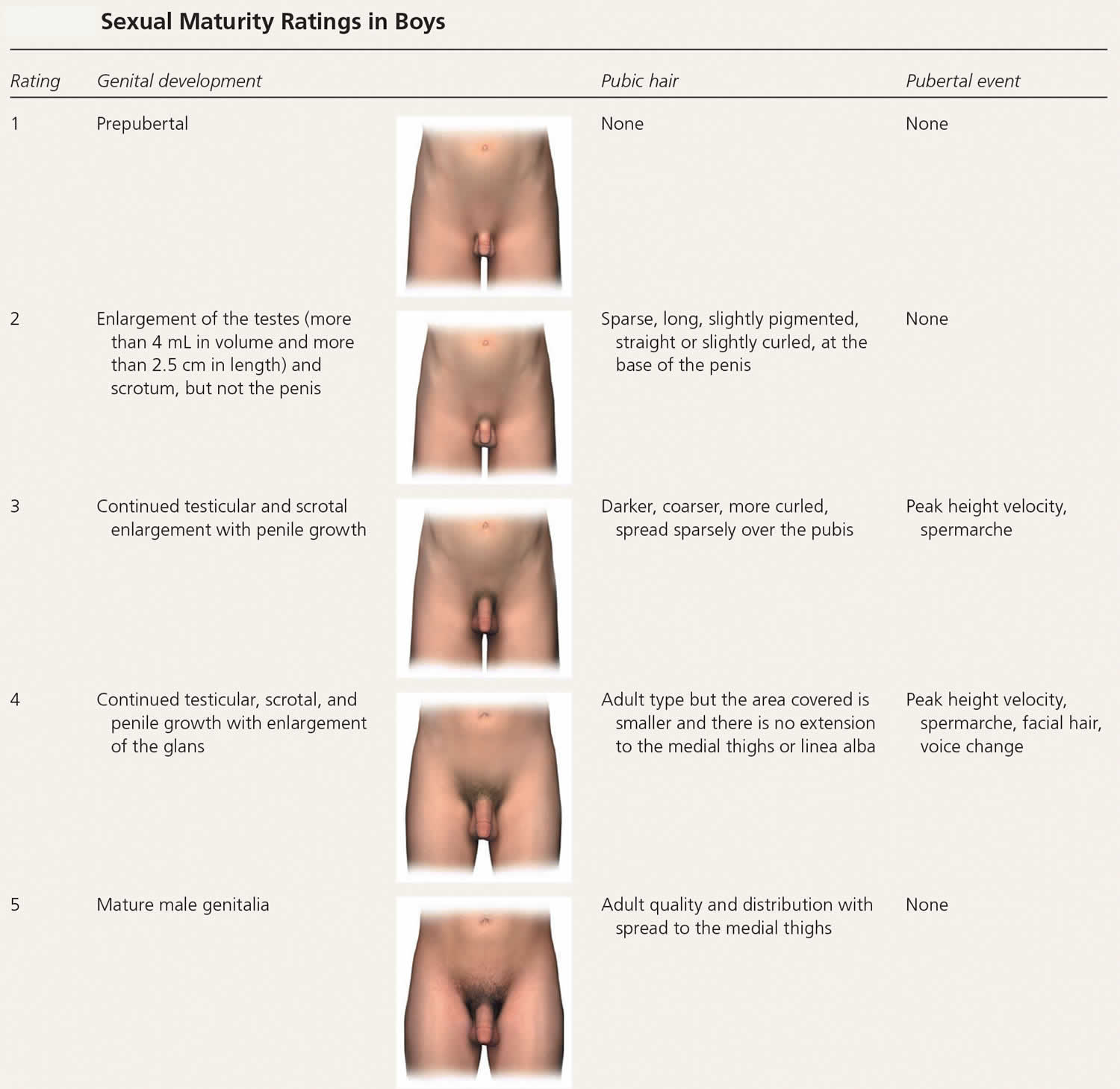
In girls, increased ovarian estradiol secretion causes breast development at a mean age of 10 years (range: eight to 12 years). Menarche typically follows 2.5 years after the onset of breast development, at an average age of 12.5 years (range: nine to 15 years) 6). In boys, testicular enlargement to at least 4 mL in volume or 2.5 cm in length is the first sign of true puberty and occurs at an average age of 11.5 years (range: 9.5 to 14 years) 7). Physical changes are described using sexual maturity ratings (Table 1 and Table 2), such as Tanner stages, and are affected by body habitus and demographic factors 8).
Linear growth velocity is about 5 cm per year from four years of age to puberty with a nadir before the pubertal growth spurt. Girls achieve peak height velocity during sexual maturity ratings 2 and 3 (mean: 8.3 cm per year, age 11 or 12 years) and boys during sexual maturity ratings 3 and 4 (mean: 9.5 cm per year, age 13 or 14 years). On average, girls complete linear growth at 15 years of age and boys at 17 years of age. After menarche, girls grow an average of 7 cm 9).
Figure 1. Normal puberty
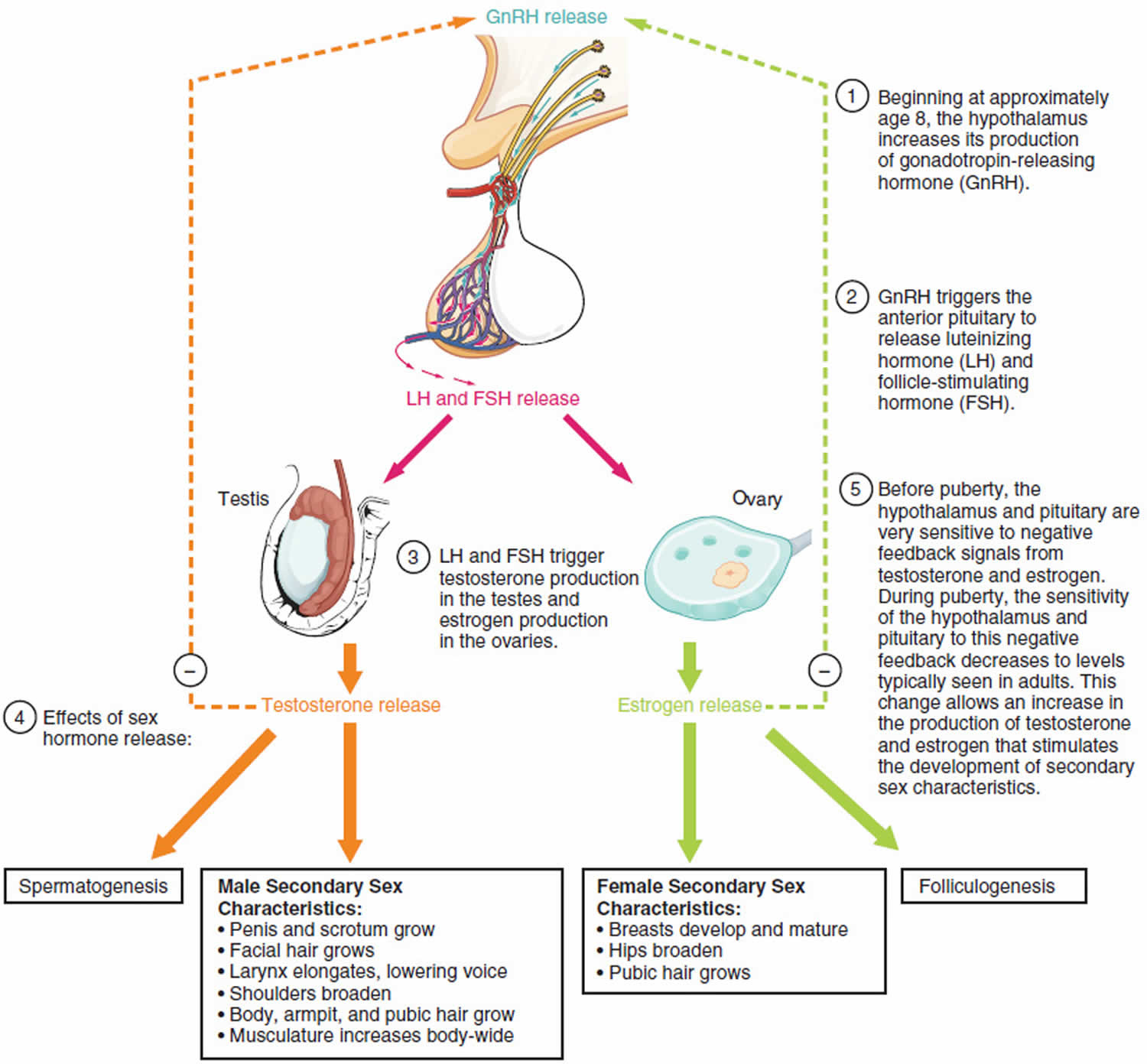
Table 1. Sexual Maturity Ratings in Girls
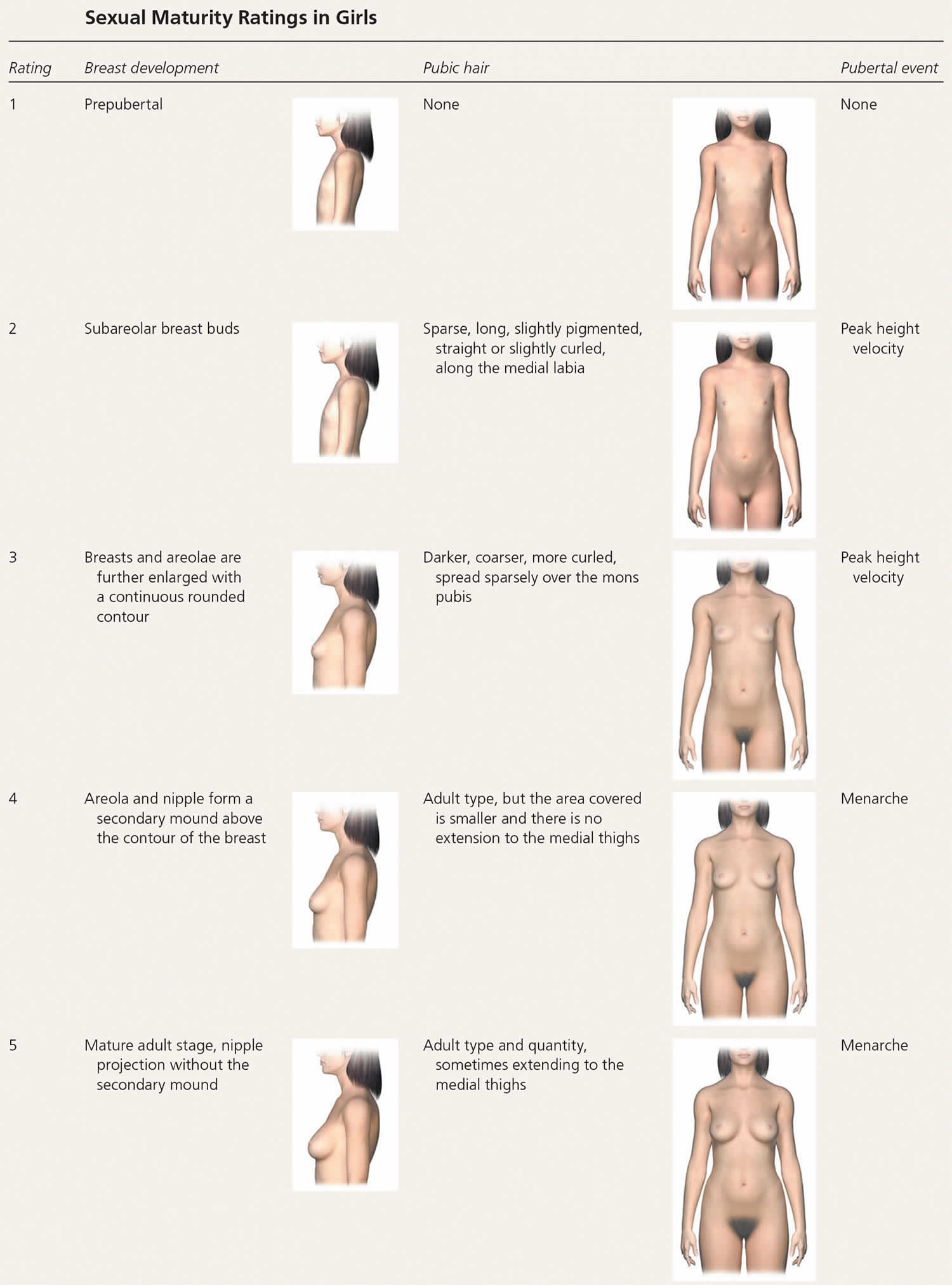
Table 2. Sexual Maturity Ratings in Boys
Delayed puberty causes
Most children with delayed puberty, especially boys, are “late bloomers” and otherwise healthy. This is called constitutional delay of growth and puberty. Usually, at least one parent was also a late bloomer. Some health problems and being underweight can cause delayed puberty. Less commonly, a problem with hormones causes it.
Puberty can be delayed in children who have not gotten proper nutrition due to long-term illnesses. Also, some young girls who undergo intense physical training for a sport, such as running or gymnastics, start puberty later than normal 12).
In other cases, the delay in puberty is not just due to slow maturation but occurs because the child has a long-term medical condition known as hypogonadism, in which the sex glands (the testes in men and the ovaries in women) produce few or no hormones.
Hypogonadism can be divided into two categories: secondary hypogonadism and primary hypogonadism.
- Primary hypogonadism also known as hypergonadotropic hypogonadism, the problem lies in the ovaries or testes, which fail to make sex hormones normally. In hypergonadotropic hypogonadism (primary hypogonadism), gonadal insufficiency delays puberty and results in elevated levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). Conditions causing hypergonadotropic hypogonadism can be congenital or acquired and are collectively more common in girls (26%) than in boys (7%) with delayed puberty 13). Some causes include 14):
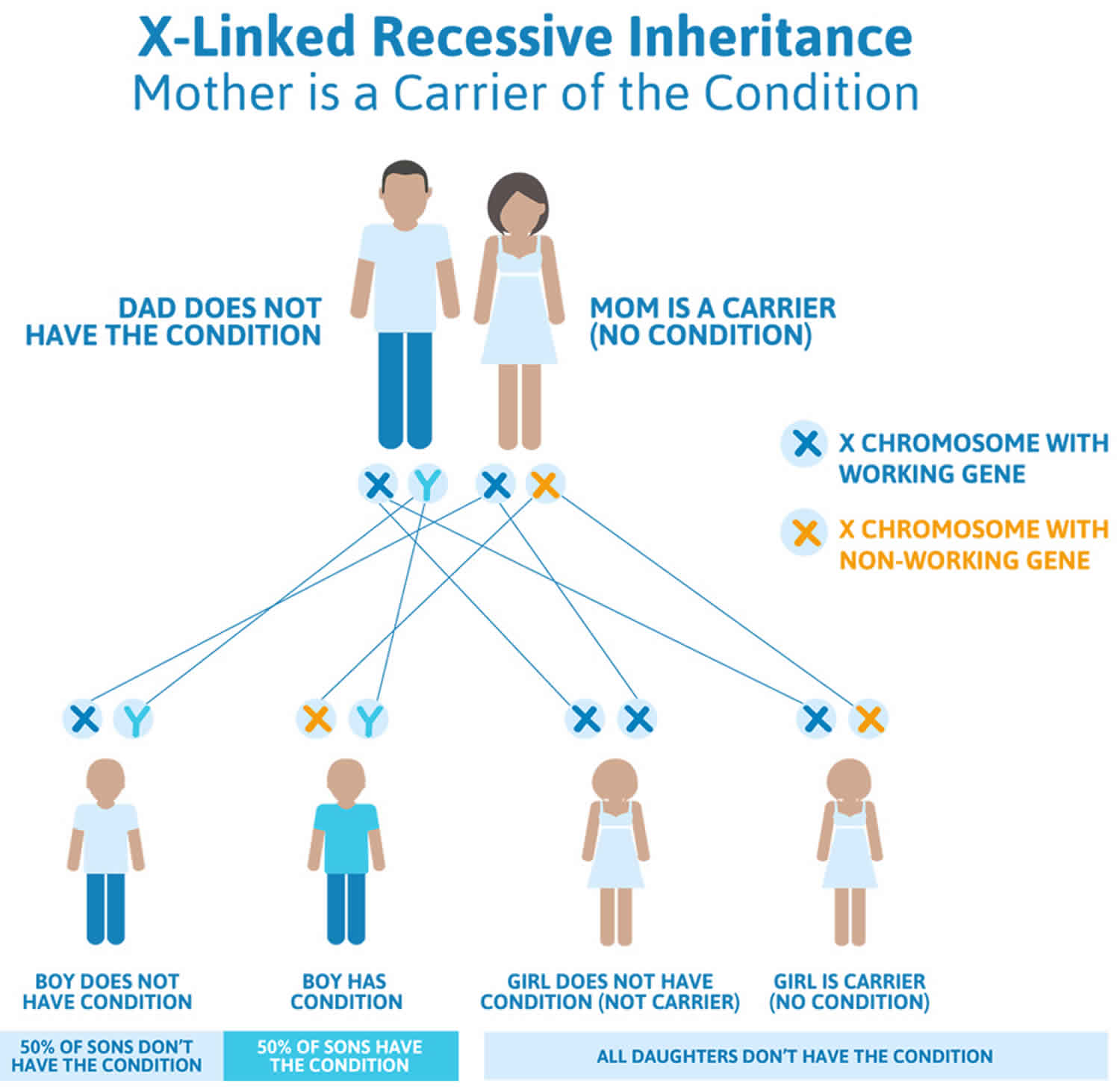
- Genetic disorders, especially Turner syndrome (in women) and Klinefelter syndrome (in men)
- Certain autoimmune disorders
- Developmental disorders
- Radiation or chemotherapy
- Infection
- Surgery
- Secondary hypogonadism also known as central hypogonadism or hypogonadotropic hypogonadism, is caused by a problem with the pituitary gland or hypothalamus (part of the brain). In secondary hypogonadism, the hypothalamus and the pituitary gland fail to signal the gonads to properly release sex hormones. Hypogonadotropic hypogonadism (secondary hypogonadism or central hypogonadism) is characterized by low levels of FSH and LH and further classified by the pathology. Causes of secondary hypogonadism include 15):
- Kallmann syndrome, a genetic problem that also diminishes the sense of smell
- Isolated hypogonadotropic hypogonadism, a genetic condition that only affects sexual development but not the sense of smell
- Prior radiation, trauma, surgery, or other injury to the brain or pituitary
- Tumors of the brain or pituitary.
Functional hypogonadotropic hypogonadism is caused by chronic disease, stress, or inadequate nutrition, and the condition may be transient or reversed. Persistent hypogonadotropic hypogonadism is caused by a congenital abnormality in the hypothalamic-pituitary-gonadal axis or an acquired etiology such as a central nervous system tumor, trauma, surgery, or radiation 16). Patients with persistent hypogonadotropic hypogonadism require treatment to induce puberty, maintain normal adult levels of sex steroids, and optimize fertility 17). Table 3 includes the differential diagnosis of delayed or absent puberty.
Table 3. Delayed or Absent Puberty differential diagnosis
| Diagnosis | Characteristics | Treatment | |
|---|---|---|---|
| Generally benign variant | |||
| Constitutional delay of growth and puberty | Normal growth velocity, history of delayed puberty in parents, delayed bone age | Surveillance every 6 months to evaluate for progression of pubertal development | |
| Functional hypogonadotropic hypogonadism * | |||
| Celiac disease | Abdominal pain, malabsorption, anemia, poor weight gain; short stature may be the only symptom; positive serology results, confirmed with endoscopic biopsy | Gluten-free diet, surveillance | |
| Diabetes mellitus | Polyuria, polydipsia, polyphagia, weight loss, or known but poorly controlled disease; confirmed by serology | Treat underlying disease | |
| Hyperthyroidism | Weight loss, heat intolerance, insomnia, tachycardia, hypertension; confirmed with serology | Treat underlying disease | |
| Hypothyroidism | Weight gain, cold intolerance, fatigue, bradycardia; confirmed with serology | Treat underlying disease | |
| Inadequate nutrition for metabolic needs (e.g., eating disorder) | Weight loss or poor weight gain, excessive exercise, food restriction, purging | Weight restoration, treatment of underlying disorder | |
| Inflammatory bowel disease | Abdominal pain, constipation, diarrhea, hematochezia, poor weight gain, elevated serum erythrocyte sedimentation rate and C-reactive protein; confirmed with endoscopic biopsy | Treat underlying disease | |
| Persistent hypogonadotropic hypogonadism | |||
| Genetic† | |||
| Congenital hypogonadotropic hypogonadism | Gonadotropin-releasing hormone deficiency, bilateral cryptorchidism, micropenis, unilateral renal agenesis, synkinesis (mirror movements), cleft lip or palate, hearing loss, dental agenesis, skeletal malformations | Referral to a pediatric endocrinologist for hormone therapy | |
| Kallmann syndrome ‡ | Anosmia in addition to congenital hypogonadotropic hypogonadism presentation | Referral to a pediatric endocrinologist for hormone therapy | |
| Acquired | |||
| Central nervous system trauma, surgery, or radiation | History of trauma, surgery, or central nervous system radiation for prior malignancy; may present similarly to central nervous system tumor if acute | Referral to a pediatric endocrinologist for hormone therapy; other referrals as necessary for treatment of underlying disease | |
| Central nervous system tumors | Headaches, vision changes, seizures, suggestive magnetic resonance imaging findings of the brain and pituitary | Referral for diagnosis and treatment of underlying disease (e.g., neurosurgeon, endocrinologist) | |
| Hypergonadotropic hypogonadism § | |||
| Chemotherapy, radiation, or trauma to gonads | History | Referral to a pediatric endocrinologist for hormone therapy | |
| Klinefelter syndrome (boys) | Tall stature, learning disabilities, relatively small testes (3 to 6 mL) for degree of androgenization; 47,XXY karyotype | Referral to a pediatric endocrinologist for hormone therapy | |
| Oophoritis or orchitis | History of mumps infection in boys | Referral to a pediatric endocrinologist for hormone therapy | |
| Turner syndrome (girls) | Short stature, facial dysmorphism, webbed neck, brachydactyly, heart defects; in cases of mosaicism, short stature may be the only sign; 45,X or related karyotype | Referral to a pediatric endocrinologist for hormone therapy and other comprehensive care | |
Footnotes:
* Other causes include cystic fibrosis, juvenile rheumatoid arthritis, systemic lupus erythematosus, sickle cell disease, thalassemia, chronic renal disease, and malnutrition.
† Other syndromes include septo-optic dysplasia, Prader-Willi, Laurence-Moon, Bardet-Biedl, and CHARGE.
‡ 50% of congenital hypogonadotropic hypogonadism cases; five times more prevalent in boys. It is caused by disrupted migration of gonadotropin-releasing hormone–secreting neurons and the olfactory bulbs.
§ Other causes include gonadal dysgenesis, premature ovarian insufficiency (often autoimmune in nature), vanishing testes syndrome, testicular biosynthetic defects, and luteinizing hormone and follicle-stimulating hormone receptor defects.
[Source 18) ]Constitutional delayed puberty
Most often, delayed puberty is a pattern of growth and development in a family. A child’s parents, uncle, aunt, brothers, sisters, or cousins might have developed later than usual too. This is called constitutional delay and usually doesn’t need any treatment. These “late bloomers” in time will develop normally, just later than most of their peers.
Constitutional delay of growth and puberty is the most common cause of delayed puberty in boys (60%) and girls (30%) 19). It represents an extreme of the normal spectrum of pubertal timing and is a diagnosis of exclusion 20). For more than 75% of patients with constitutional delay of growth and puberty, family history may reveal parental pubertal delay 21).
Medical problems
Some medical problems can cause delays in puberty:
- Some kids and teens with chronic illnesses like diabetes, cystic fibrosis, kidney disease, or even asthma may go through puberty at an older age. That’s because their illnesses can make it harder for their bodies to grow and develop. Proper treatment and better control of these conditions can help make delayed puberty less likely.
- Being malnourished — not eating enough food or not getting good nutrients — can make someone develop later than peers who eat a healthy, balanced diet. This can happen because of food insecurity, as well as disordered eating or excess physical activity. Teens with the eating disorder anorexia nervosa, for example, often lose so much weight that their bodies can’t develop properly. Girls who are extremely active in sports may be late developers because their level of exercise keeps them so lean. Girls’ bodies need enough fat before they can go through puberty or get their periods.
- Problems in the pituitary gland or thyroid gland, which make hormones important for body growth and development, also can delay puberty.
- Chromosome disorders can delay puberty in some people. Chromosomes are made up of DNA that contain our body’s construction plans. So when they have problems, it can affect normal growth processes. For example:
- Turner syndrome is when one of a female’s two X chromosomes is abnormal or missing. This causes problems with how her body grows and makes sex hormones, and how her ovaries develop. Women who have Turner syndrome are shorter than normal, may not go through puberty in the usual way, and may have other medical problems. Sometimes, puberty starts at a normal time, and then stalls or stops after a few years.
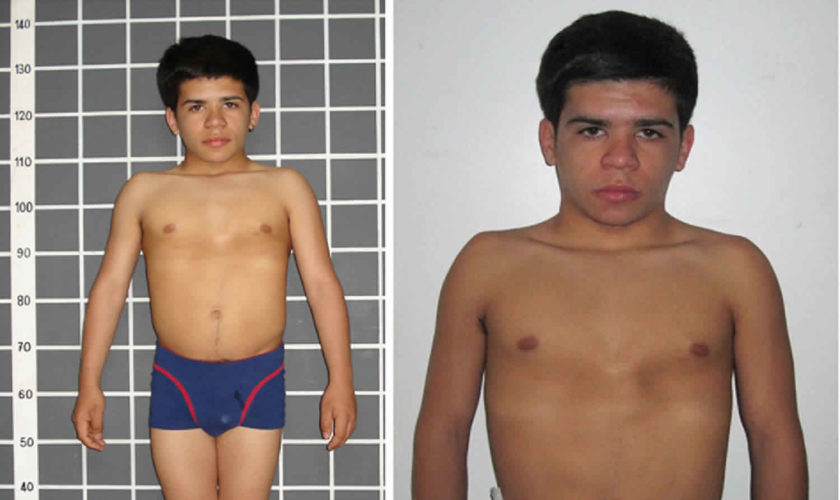
- Klinefelter syndrome is when males are born with an extra X chromosome (XXY instead of XY). This condition can affect testicular function and sexual development. These boys usually are tall for their age, might have learning problems, and may have other medical problems. Puberty usually starts at a normal time, but then stalls.
Delayed puberty signs and symptoms
Lacking signs of puberty is the primary indicator that a child may be experiencing delayed puberty. The following are the most common symptoms of delayed puberty. However, each child may experience symptoms differently. The symptoms of delayed puberty may resemble other problems or medical conditions. Always consult your child’s doctor for a diagnosis.
Symptoms of delayed puberty may include:
Delayed puberty girls
- Lack of any breast development by age 13
- More than four years between initial breast growth and first menstrual period
- Failure to menstruate by age 14
Delayed puberty boys
- Lack of testicular enlargement by age 14
- Lack of pubic hair by age 15
- More than four years to complete adult genital development
Delayed puberty diagnosis
If a boy or girl hasn’t shown signs of puberty as they move into the teen years, doctors will:
- Do an exam.
- Take a medical history, including whether others in the child’s family had a similar growth pattern.
- Ask about any medicines the child takes.
- Check the growth chart to see if there’s a pattern that points to a problem.
Figure 2. Diagnostic approach to delayed puberty or absent pubertal development
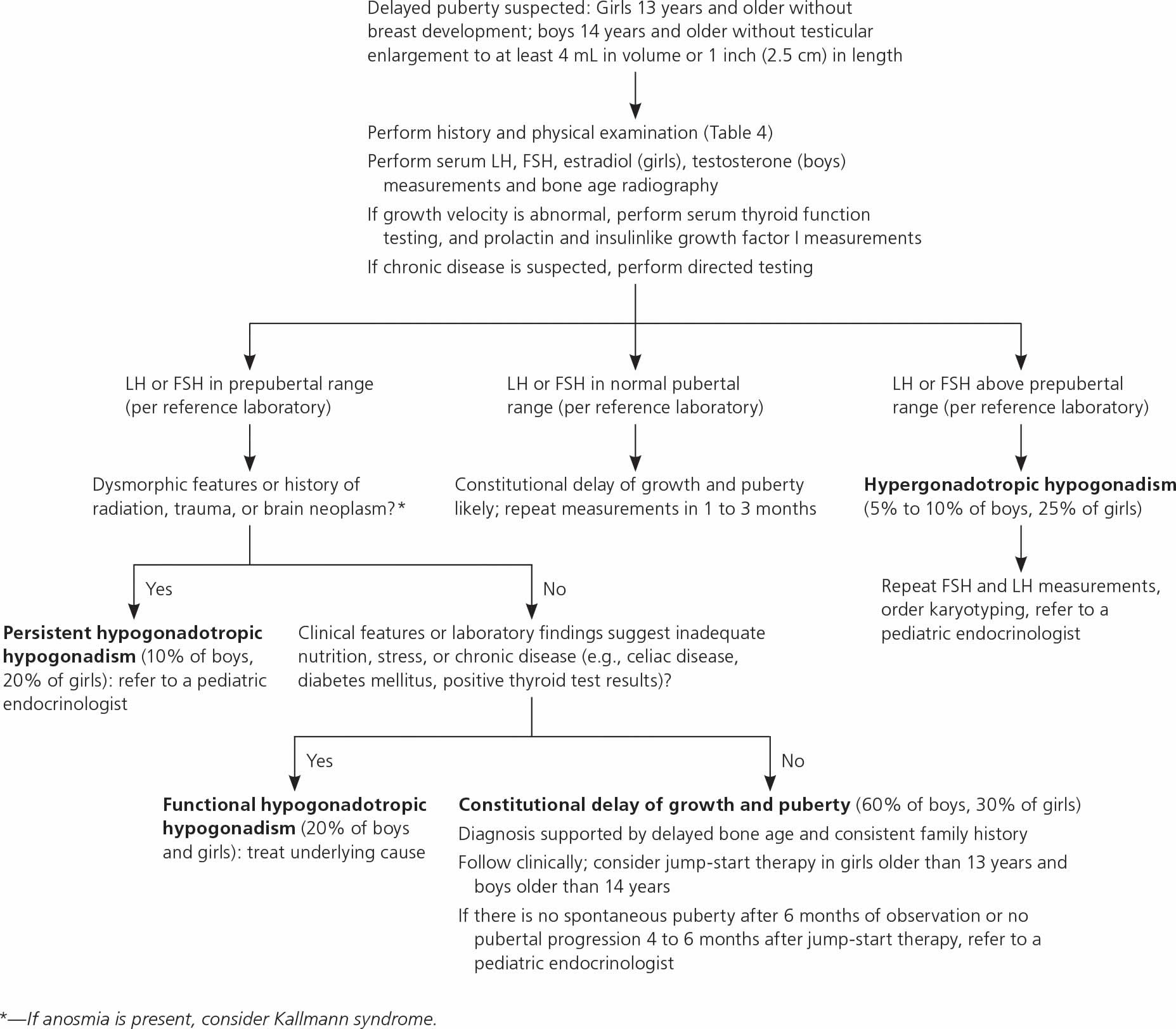
Table 4. Delayed puberty history and physical examination findings
| Findings | Possible diagnoses |
|---|---|
| Abdominal pain | Gastrointestinal disease |
| Anosmia | Kallmann syndrome |
| Asymmetric testes | Oophoritis or orchitis |
| Body mass index and weight (on growth charts) | Low: eating disorder, caloric insufficiency, gastrointestinal or other systemic disease |
| Chemotherapy, radiation treatment, brain tumor | Hypogonadism |
| Cryptorchidism or orchidopexy | Hypogonadism |
| Dysmorphic features (webbed neck, short stature, low hairline) | Turner syndrome |
| Enlarged thyroid | Hypothyroidism |
| Family history of late puberty | Constitutional delay of growth and puberty |
| Galactorrhea | Hyperprolactinemia |
| Growth velocity | Peripubertal growth slowing, pathologic growth due to underlying condition |
| Height (growth chart) | Short stature: Turner syndrome, constitutional delay of growth and puberty |
| Tall stature: Klinefelter syndrome | |
| Joint pain | Inflammatory disorder |
| Neurologic assessment (abnormal examination findings or symptoms such as headaches, vision changes) | Intracranial pathology |
| Red (vs. dull pink) or thin vaginal mucosa | Lack of estrogen exposure (hypogonadism) |
| Sexual maturity rating | Delayed pubertal development (unspecified) |
| Small, firm testes | Klinefelter syndrome |
| Temperature intolerance, gastrointestinal symptoms, tremor, depression, palpitations | Thyroid disease |
| Trauma (head) | Hypogonadism |
| Vasomotor symptoms in girls | Ovarian insufficiency |
| Weight loss, stress, excessive exercise, inadequate nutrition, fatigue | Eating disorder, caloric insufficiency |
In addition to a complete medical history and physical examination, diagnosis of delayed puberty may include:
- Blood tests. Tests to check for chromosomal abnormalities, to measure hormone levels, and to test for diabetes, thyroid, pituitary, chromosomal, anemia, and other conditions that may delay puberty.
- X-ray. A diagnostic test that uses invisible electromagnetic energy beams to produce images of internal tissues, bones, and organs onto film. Simple X-ray of the left hand and wrist bones is often performed to determine bone maturity.
- Computed tomography scan (also called a CT or CAT scan). A diagnostic imaging procedure that uses a combination of X-rays and computer technology to produce horizontal, or axial, images (often called slices) of the body. A CT scan shows detailed images of any part of the body, including the bones, muscles, fat, and organs. CT scans are more detailed than general X-rays.
- Magnetic resonance imaging (MRI). A diagnostic procedure that uses a combination of large magnets, radiofrequencies, and a computer to produce detailed images of organs and structures within the body.
If doctors find a problem, they usually refer families to a pediatric endocrinologist, a doctor who specializes in treating kids and teens who have growth problems, or to another specialist for more tests or treatment.
Initial workup should include measurements of serum FSH, LH, testosterone in boys or estradiol in girls, and bone age radiography (Table 5). If abnormal growth velocity is a concern, serum thyroid function, prolactin, and insulin like growth factor I should be assessed 24). Constitutional delay of growth and puberty can be difficult to distinguish from persistent hypogonadotropic hypogonadism; the latter may be diagnosed at 18 years of age if there is inadequate response to jump-start therapy (which is defined later in this section), and sex steroid replacement is still required 25).
Bone age indicates the degree of sex steroid effect on bone maturation and future growth potential 26). For example, patients with constitutional delay of growth and puberty generally have a delay of more than two years, but this finding is nonspecific 27).
Often, the doctor will find no underlying physical problem. Most kids with delayed puberty are just developing a bit later than average and will catch up.
Table 5. Diagnostic testing in the evaluation of pubertal disorders
| Test | Condition-specific findings
|
||
|---|---|---|---|
| Precocious puberty | Delayed puberty | ||
| Laboratory testing (refer to local reference values) | |||
| First-line | |||
| Serum estradiol | Elevated (girls): estrogen exposure; if markedly elevated (> 100 pg per mL [367 pmol per L]), evaluate for ovarian tumor, especially if luteinizing hormone is suppressed | Low (girls): prepubertal, may suggest poor ovarian function in response to gonadotropins | |
| Serum testosterone | Elevated: testicular (boys), adrenal, or exogenous source | Low (boys): prepubertal, poor response of testes to gonadotropin stimulation | |
| Serum LH and follicle-stimulating hormone | Prepubertal levels: benign variant or peripheral precocious puberty | High: gonadal insufficiency, Turner syndrome, Klinefelter syndrome | |
| Postpubertal levels > 0.3 mIU per mL (0.3 IU per L): central precocious puberty | Low: hypogonadotropic hypogonadism, constitutional delay of growth and puberty | ||
| If indicated | |||
| Directed testing (e.g., for celiac disease; diabetes mellitus; or hepatic, renal, or inflammatory conditions) | — | Functional hypogonadotropic hypogonadism, seek underlying cause | |
| Gonadotropin-releasing hormone analogue stimulation test | Elevated LH: central precocious puberty (vs. benign variant) in complex clinical scenarios | Used in complex clinical scenarios | |
| Suppressed LH but elevated sex steroids: peripheral precocious puberty | |||
| Karyotyping | — | Turner syndrome, Klinefelter syndrome | |
| Serum 17-hydroxyprogesterone | Elevated: nonclassic (late onset) congenital adrenal hyperplasia | — | |
| Serum dehydroepiandrosterone sulfate | Elevated: adrenal source, premature adrenarche (mild elevation) vs. peripheral precocious puberty | Normal for age: may suggest persistent hypogonadotropic hypogonadism rather than constitutional delay of growth and puberty | |
| Serum human chorionic gonadotropin (boys) | Elevated: human chorionic gonadotropin–secreting germ cell tumor | — | |
| Serum insulinlike growth factor I | — | Low: growth hormone deficiency (if low for both bone and chronologic age) | |
| Serum prolactin | — | High: prolactin-secreting tumor, hypothyroidism, other neoplasm | |
| Serum thyroid-stimulating hormone and free thyroxine | Thyroid disease | Thyroid disease | |
| Imaging | |||
| First-line | |||
| Bone age radiography | Advanced (> 2 standard deviations): more likely to be central or peripheral precocious puberty, less likely to be benign pubertal variant | Delayed: constitutional delay of growth and puberty, underlying chronic disease | |
| If indicated | |||
| Adrenal imaging | Adrenal tumor | — | |
| Magnetic resonance imaging (brain and pituitary) | Central nervous system lesion | Central nervous system lesion | |
| Pelvic or testicular ultrasonography | Ovarian or testicular tumor; greater ovarian volume may indicate central precocious puberty (vs. benign variant) | Absence of the uterus (e.g., androgen insensitivity, Müllerian system abnormalities) | |
Delayed puberty treatment
Specific treatment for delayed puberty will be determined by your child’s doctor based on:
- Your child’s age, overall health, and medical history
- Extent of the condition
- Your child’s tolerance for specific medications, procedures, or therapies
- Expectations for the course of the condition
- Your opinion or preference
Treatment for delayed puberty depends on the cause of the problem. Often, when the underlying cause is treated, puberty proceeds normally.
Many children with delayed puberty will eventually go through an otherwise normal puberty, just at a late age. If the delayed puberty is due to heredity, no treatment is usually necessary. Some late bloomers struggle with waiting for the changes of puberty to start. So doctors may offer hormone treatment:
- Boys might get a short course of treatment with testosterone (usually a monthly injection for 4–6 months) to get the changes of puberty started.
- Girls might get low doses of estrogens for 4–6 months to start breast development.
Girls older than 13 years and boys older than 14 years with possible constitutional delay of growth and puberty or gonadotropin-releasing hormone deficiency may be offered jump-start therapy to induce puberty 29). For example, treating boys with testosterone cypionate or enanthate (e.g., 50 to 100 mg intramuscularly per month) and girls with overnight transdermal estradiol (e.g., 6.2 mcg, one-fourth of the 25-mcg 24-hour patch) for three to six months may accelerate attainment of final adult height and generally does not lead to premature epiphysis closure 30). After treatment ends, the teen’s own hormones usually take over to complete the process of puberty. If they don’t, the doctor will discuss long-term sex hormone replacement. If pubertal progression does not occur within four to six months after completing therapy, further evaluation for persistent hypogonadotropic hypogonadism and long-term hormone therapy should be initiated 31).
In some cases, surgery is necessary to correct an anatomical problem.
Other children have a long-lasting condition known as hypogonadism in which the sex glands (the testes in men and the ovaries in women) produce few or no hormones. For example, hypogonadism can occur in girls with Turner syndrome or in individuals with hypogonadotropic hypogonadism, which occurs when the hypothalamus produces little to no gonadotropin-releasing hormone (GnRH).
References [ + ]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}