



Goldenhar syndrome
Goldenhar syndrome also known as oculoauicular dysplasia, is a condition that is present at birth (congenital birth defect) arising from the abnormal development of the first and second branchial arches and mainly affects the development of the eye, ear, and spine. Goldenhar syndrome main ocular manifestation is limbal dermoid, which is found in 30%–60% of cases and is a cause of astigmatism, amblyopia, and strabismus 1).
Goldenhar syndrome usually affects one side of the face only. Characteristics include 2):
- a partially formed or totally absent ear (microtia)
- the chin may be closer to the affected ear
- one corner of the mouth may be higher than the other
- benign growths of the eye
- a missing eye.
The main sign and symptoms are facial asymmetry (one side of the face is different from the other), a partially formed ear (microtia) or totally absent ear (anotia), noncancerous (benign) growths of the eye (ocular dermoid cysts), and spinal abnormalities. Goldenhar syndrome may also affect the heart, lungs, kidneys, and central nervous system 3). The first case of Goldenhar syndrome was described in the following case report published in 1952 4). Goldenhar syndrome is due to problems that occur when the fetus is forming within the womb of the mother, in structures known as the first and second brachial arch. These structures will develop to form the neck and the head. The cause of Goldenhar syndrome is still unknown 5). Goldenhar syndrome is part of a group of conditions known as craniofacial microsomia. It is not known whether the conditions included in the group really are different conditions or part of the same problem with different degrees of severity. Treatment is age-dependent, with interventions at appropriate stages during the growth and development of the skull and face 6).
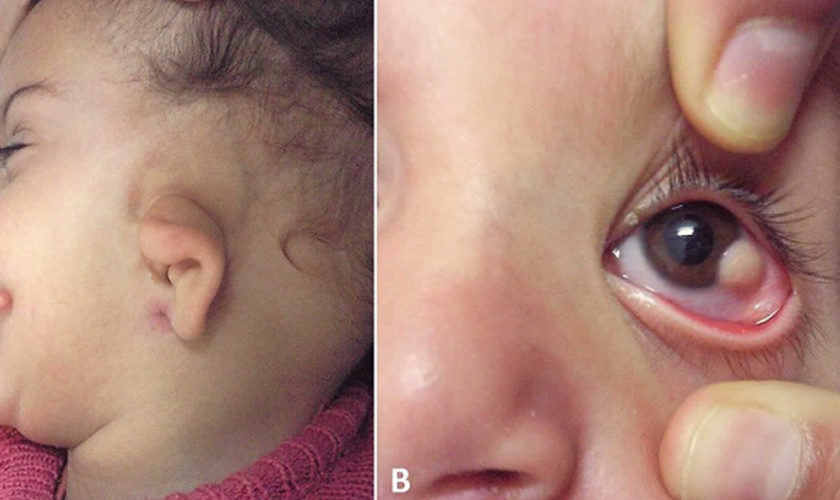
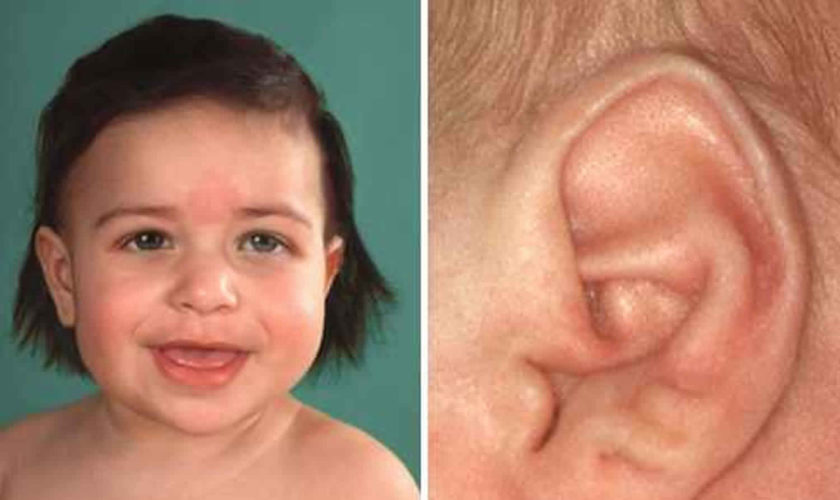
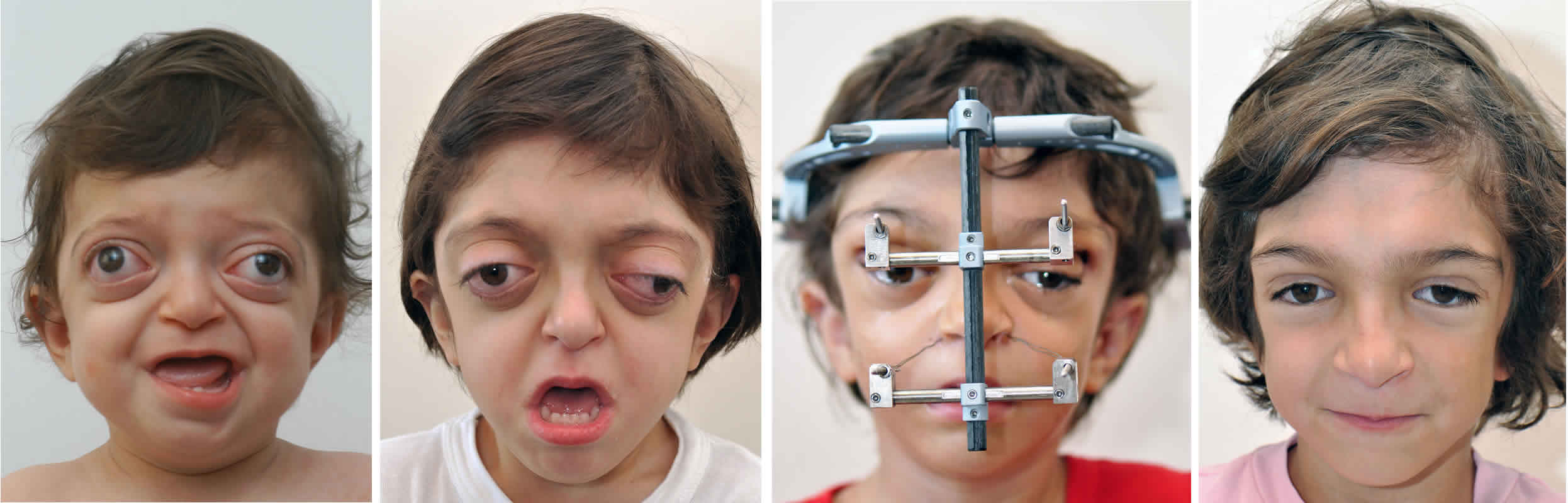
Figure 1. Goldenhar syndrome
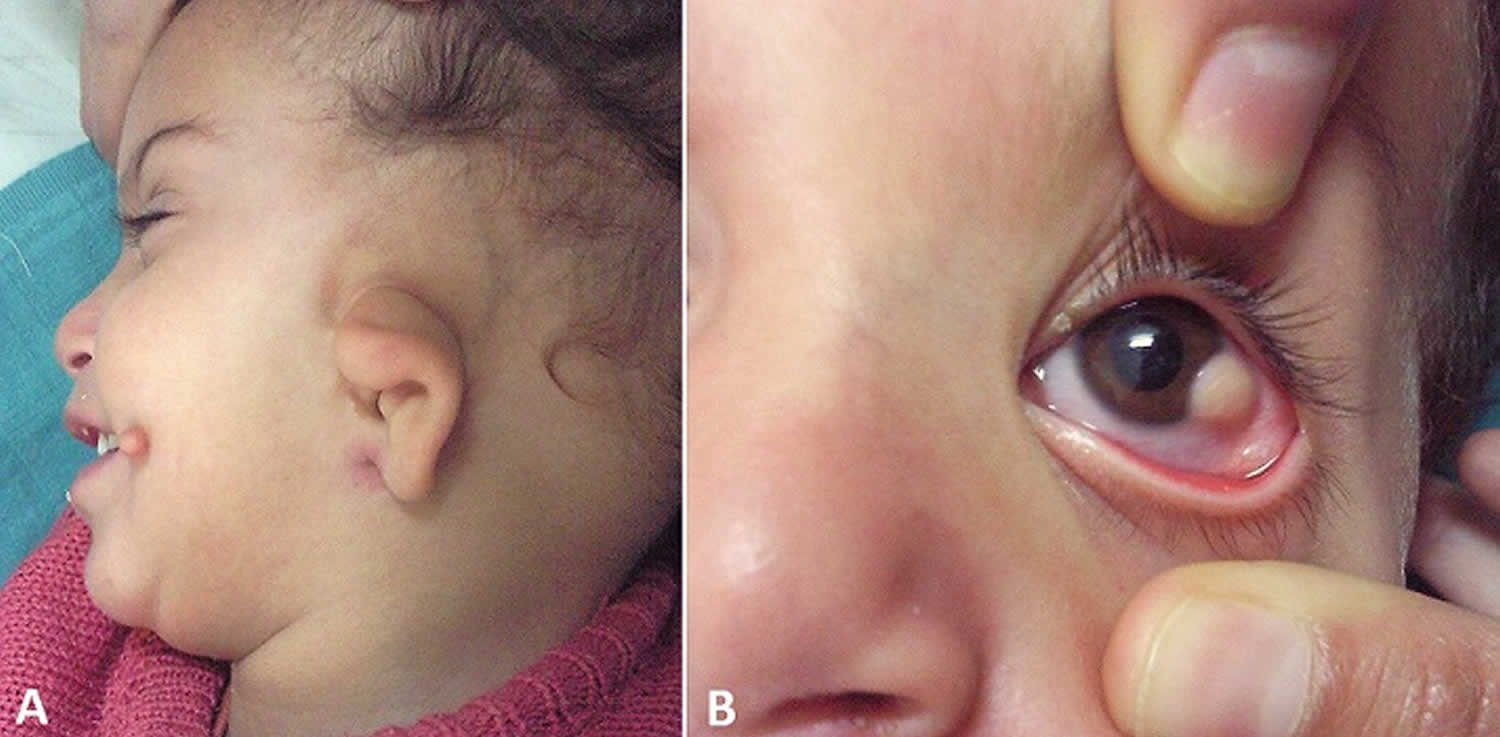
Footnote: (A) Microtia and preauricular skin tag; (B) limbal dermoid in the same child.
Will Goldenhar syndrome happen to children I have in the future?
The chances of having another child with Goldenhar syndrome is less than 1% or less. Your child has about a 3% chance of passing it on to his or her children 7).
How many people have Goldenhar syndrome and bilateral Goldenhar syndrome in the United States and worldwide?
Unfortunately, for rare diseases, there is often not a calculated incidence or prevalence because there is no official method for tracking them. However, it has been estimated that the frequency of Goldenhar disease ranges between 1 case per 3,500 births and 1 case per 25,000 births 8). Given these data, a very rough estimate for the number of people in the United States with Goldenhar syndrome ranges from 13,000-56,000 and estimates for the number of people worldwide with the syndrome ranges from 300,000-1,300,000. Currently, scientists are unaware of estimates regarding bilateral (affecting both sides of the body) disease, specifically.
Is there any research or information on people with Goldenhar syndrome having mental illness or multiple personalities?
There is one case report of a single patient diagnosed with Goldenhar syndrome who had schizophreniform disorder 9). Case reports document clinic findings associated with individual cases. It is important to keep in mind that the clinical findings documented in these case reports are based on specific individuals and may differ from one affected person to another.
Goldenhar syndrome causes
The underlying cause of Goldenhar syndrome is poorly understood. Most cases occur sporadically with no apparent explanation. Some researchers suspect that problems with blood flow or other disruptions during fetal development may contribute to the development of the condition 10). Animal models demonstrate that vascular disruption and hematoma formation affect the developing structures of the jaw and the ear regions in utero. Other hypotheses suggest that disturbance of the population of the neural crest cells occurs at 30-45 days’ gestation.
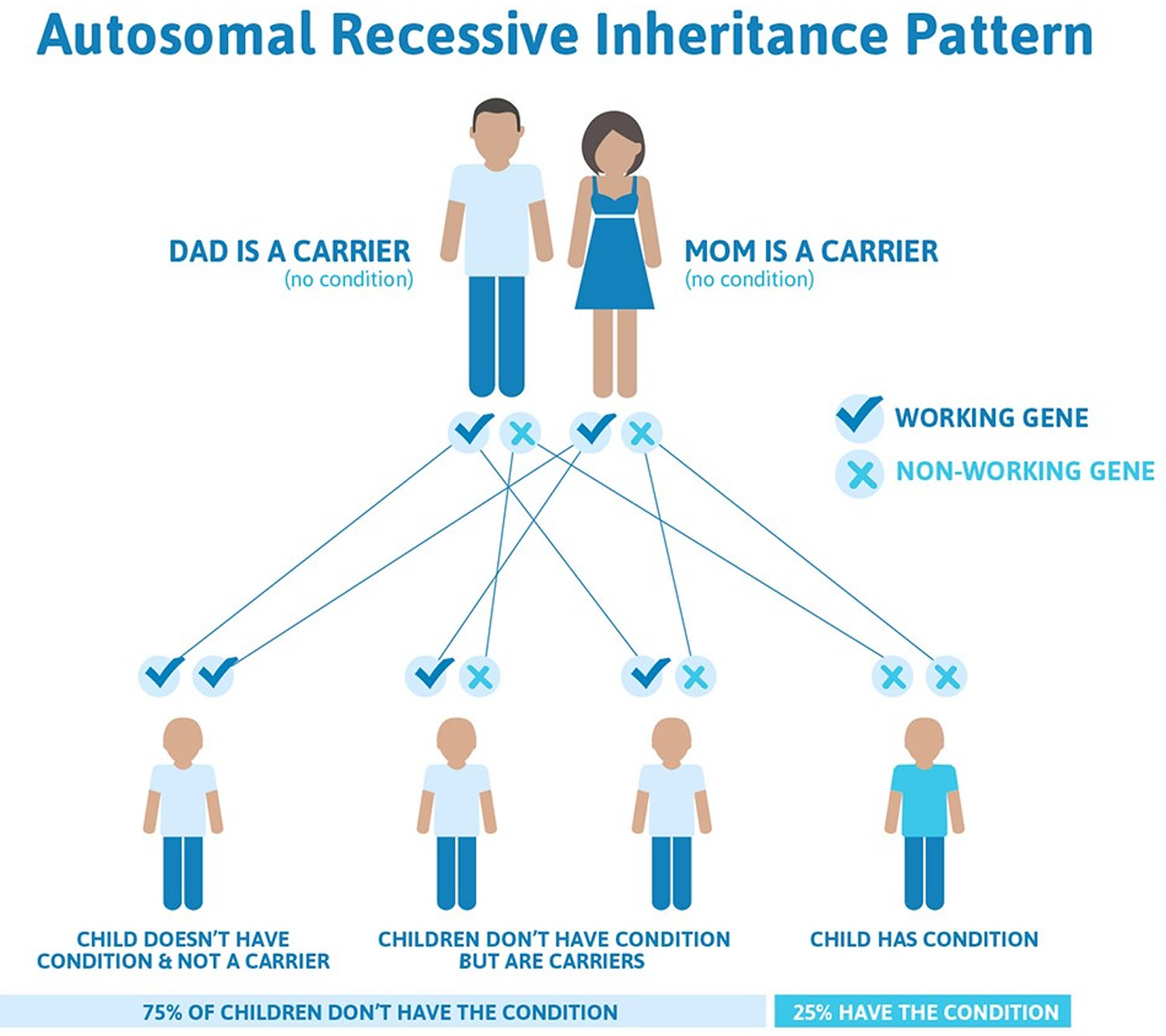
Approximately 1-2% of affected people have other family members with the condition, which suggests that genes may play a role in some cases 11).
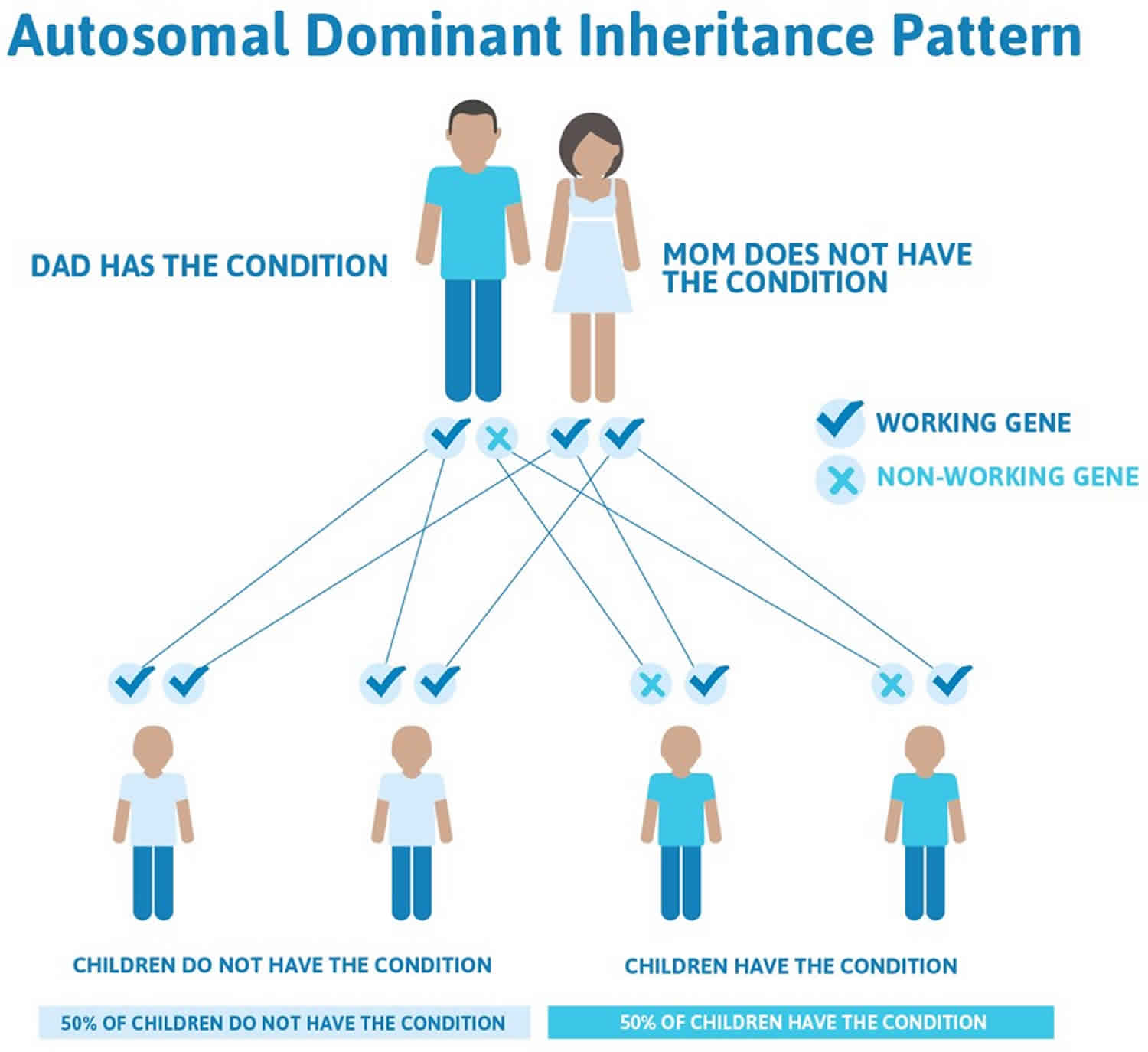
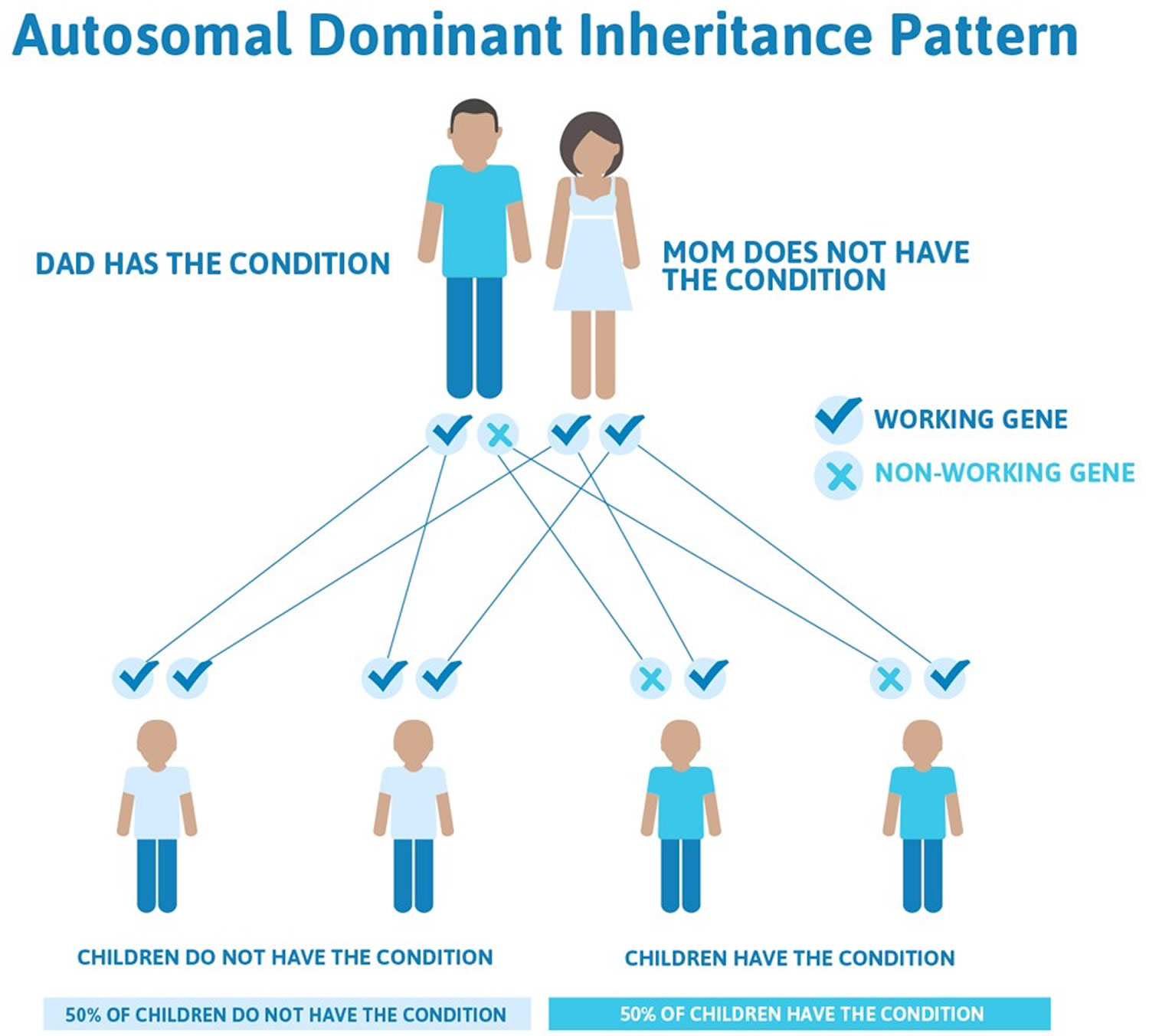
Goldenhar syndrome inheritance pattern
Most cases of Goldenhar syndrome occur sporadically in people with no family history of the condition 12). Rarely (approximately 1-2% of affected people), more than one family member can be affected. In these cases, the condition appears to be passed down through the family in an autosomal dominant manner 13).
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
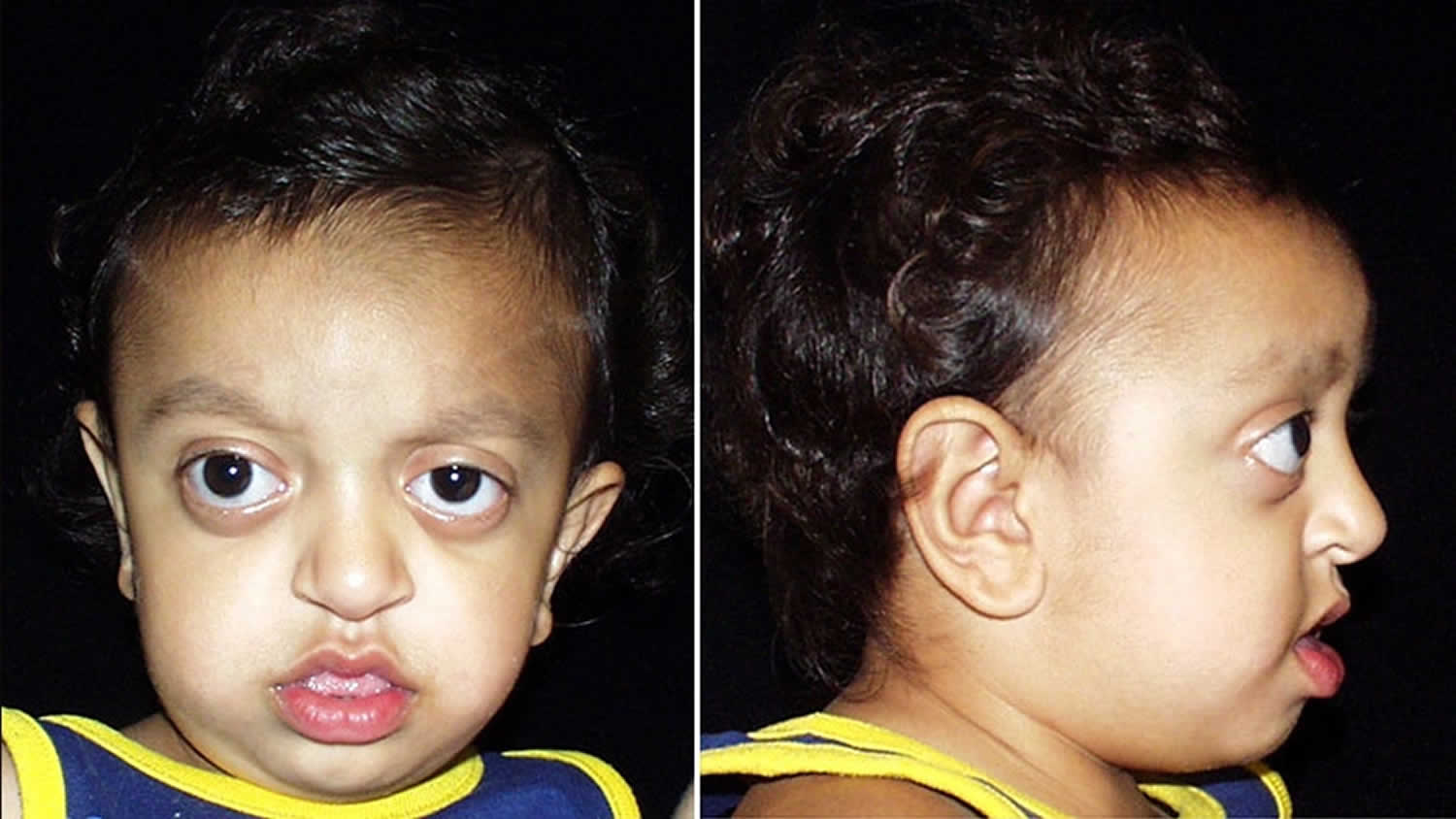
Goldenhar syndrome symptoms
The signs and symptoms of Goldenhar syndrome vary significantly from person to person. Common signs and symptoms of the condition include 14):
- Microtia (a partially formed or completely absent ear) and other ear abnormalities
- Underdeveloped facial muscles which may be associated with weakness
- Underdeveloped jaw, cheekbone and/or temple bone
- Cleft lip and/or palate
- Abnormalities of the eyes, such as anophthalmia/microphthalmia, epibulbar tumors (noncancerous growths in the eyes), retinal abnormalities, and vision loss
- An unusually large or small mouth
- Dental abnormalities
Goldenhar syndrome deformities divided into groups according to the region they affect 15):
- Ocular symptoms: Epibulbar dermoids, microphthalmia, anophthalmia, eyes asymmetry/dysmorphy, cleft eyelid, exophthalmia, strabismus
- Auricular symptoms: dacryocystitis, preauricular appendages, preauricular fistulas, ear asymmetry, microtia, atresia of the externalauditory canal
- Craniofacial deformities: cleft face, cleftlip, cleft palate, macrostomia, bifidtongue, hypoplasia of the mandible, hypoplasia of the maxilla, asymmetry of the mandible and maxilla, malocclusion, tooth discrepancies, agenesis of third molars and second premolars, supernumerary teeth, enamel and dentinmal formations, delay in tooth development
- Skeletal abnormalities: cleft spine, microcephaly, dolichocephaly, plagiocephaly, vertebral defects
- Abnormalities of internalorgans:
- heart (atrial and ventricularseptal defects, conotruncal defects, out flow tract abnormalities, persistent truncusarteriosus);
- kidneys (ectopic and/or fused kidneys, renalagenesis, multicystic kidney);
- central nervous system (diffuse cerebralhypoplasia, dilated lateral cerebral ventricles or asymptomatic hydrocephalus, corpus callosum dysgenesis, frontal hypodensities, microcephaly, asymmetrical lateral ventricles, hydrocephalus due to aqueduct of Sylvius stenosis, corpus callosum lipoma, absence of septumpellucidum, diffuse cerebralhypodensity, hypothalamichamartoma).
In most cases, only one side of the face is affected, although approximately 10-33% of people with the condition have bilateral (both sides) involvement 16).
Some people with Goldenhar syndrome may also experience hearing loss; hydrocephalus (with or without intellectual disability); heart, kidneys, and lung problems; spinal abnormalities; and/or limb malformations 17).
Goldenhar syndrome diagnosis
A diagnosis of Goldenhar disease is based on the presence of characteristic signs and symptoms. These clinical features may be observed on physical examination or may require specialized testing such as imaging studies (i.e. CT scan, X-ray, echocardiogram, ultrasound). Additional testing including certain genetic tests may also be recommended to rule out conditions that are associated with similar features 18).
Goldenhar syndrome treatment
The treatment of Goldenhar syndrome is based on the signs and symptoms present in each person. Ideally, affected children should be managed by an experienced multidisciplinary craniofacial team. Treatment is age dependent and certain interventions may be recommended at different stages of growth and development 19).
The following are examples of medical issues that may need to be addressed in a person affected by Goldenhar syndrome 20):
- Feeding issues – some people affected by Goldenhar syndrome may have feeding difficulties caused by the associated craniofacial abnormalities. Interventions may include special bottles, supplemental nasogastric feedings, and gastrostomy tube placement.
- Breathing problems – affected people with an underdeveloped lower jaw may have difficulty breathing or develop sleep apnea. In these cases, referral to appropriate medical specialists is recommended so appropriate care can be provided.
- Hearing loss – a hearing evaluation is recommended in all children with Goldenhar syndrome by 6 months of age. In those with hearing impairment, hearing aids or other treatments may be recommended.
- Epibulbar tumors (noncancerous growths in the eyes) – these tumors may need to be surgically removed if they are particularly large or interfere with vision.
- Craniofacial abnormalities (i.e. cleft lip and/or palate), congenital heart defects, kidney problems, and/or spine abnormalities – some of the characteristic symptoms associated with Goldenhar syndrome may require surgical repair.
- Speech – people affected by Goldenhar syndrome are at an increased risk for a variety of speech problems due to the many associated craniofacial abnormalities. A speech evaluation and/or speech therapy may, therefore, be recommended in some affected people.
Will my child need surgery?
Depending on the severity of Goldenhar syndrome, your child may have some or all of the following surgeries:
- lowering of the jaw on the affected side
- lengthening of the lower jaw
- 3 to 4 operations to rebuild the outer ear
- addition of bone to build up the cheeks
- soft tissue may need to be added to the face
New advances in procedures to correct the symptoms of Goldenhar syndrome are constantly being developed.
References [ + ]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}