




Urinary frequency
Urinary frequency is the need to urinate many times during the day, at night (nocturia), or both but in normal or less-than-normal volumes 1). Urinary frequency may be accompanied by a sensation of an urgent need to void (urinary urgency). How often you have to urinate is a good indicator of your body’s overall state of hydration. Most people urinate about 4 to 8 times a day, mostly in the daytime. Normally, adults pass between 3 cups (700 milliliters) and 3 quarts (3 liters) of urine a day. Urinary frequency is distinguished from polyuria, which is urine output of >3 L/day.
If you’re going more often than that, it could simply mean that you may be drinking too much fluid or consuming too much caffeine, which is a diuretic and flushes liquids out of the body.
Excessive urination can refer to:
- An increased volume of urine (polyuria)
- A normal volume of urine with the need to go more often (urinary frequency)
- Both an increased volume of urine (polyuria) and normal volume of urine with the need to go more often (urinary frequency)
Many people particularly notice polyuria (urine output of >3 L/day) because they have to get up to urinate during the night (nocturia). Nocturia also can occur if people drink too much fluid too close to bedtime, even if they drink no more than normal overall.
Urinary frequency can be a sign of several more serious conditions, frequent urination usually results from disorders of the lower genitourinary tract such a bladder infection, interstitial cystitis also called painful bladder syndrome, which is a chronic inflammatory disorder of the bladder or prostate problems. Inflammation of the bladder, urethra, or both causes a sensation of the need to urinate. However, this sensation is not relieved by emptying the bladder, so once the bladder is emptied, patients continue trying to void but pass only small volumes of urine.
Frequent urination also can be a sign of a heart condition with leg swelling or a symptom of an overactive bladder, a common, easily treated condition that could be caused by several problems, including nerve damage, medications, infections, being overweight and estrogen deficiency.
If you’re a woman, the need to urinate frequently also may be a sign of poorly supported pelvic organs, such as the bladder. This is when the bladder drops into the vaginal opening because of weak pelvic floor muscles, typically following childbirth.
Some people find they need to urinate more frequently at night as they get older. That’s fairly typical, but most people after the age of 60 rarely get up more than twice a night, so more than that can be related to an overall indication of your health.
Depending on what’s causing your frequent urination, you may experience other urinary problems, such as:
- Pain or discomfort during urination
- A strong urge to urinate
- Difficulty urinating
- Loss of bladder control
- Unusual urine color
Several factors may be linked to frequent urination, such as:
- Infection, disease, injury or irritation of the bladder
- Conditions that increase urine production
- Changes in muscles, nerves or other tissues affecting bladder function
- Certain cancer treatments
- Drugs or beverages that increase urine production
Specific diseases, conditions or other causes of frequent urination include:
- Anterior prolapse (cystocele)
- Anxiety disorders
- Benign prostatic hyperplasia (BPH)
- Bladder stones
- Change in kidney function
- Diabetes insipidus
- Diuretics (water retention relievers)
- Excess consumption of total fluids, alcohol or caffeine
- Interstitial cystitis (also called painful bladder syndrome)
- Kidney infection (pyelonephritis)
- Overactive bladder
- Pregnancy
- Prostatitis
- Radiation treatment affecting the pelvis or lower abdomen
- Type 1 diabetes
- Type 2 diabetes
- Urethral stricture (narrowing of the urethra)
- Urinary incontinence
- Urinary tract infection (UTI)
- Vaginitis
Urinary frequency key points
- Urinary tract infection (UTI) is the most common cause in children and women.
- Urinary frequency in elderly men is often caused by bladder neck obstruction secondary to prostate enlargement or cancer. These patients usually require postvoid residual urine volume determination.
- Prostate disease is a common cause in men aged > 50 years.
- Excessive intake of caffeine can cause urinary frequency in healthy people.
Make an appointment with your doctor if you’re urinating more frequently than usual and if:
- There’s no apparent cause, such as drinking more total fluids, alcohol or caffeine
- The problem disrupts your sleep or everyday activities
- You have other urinary problems or worrisome symptoms
Warning signs
See your doctor as soon as possible if you have frequent urination along with any of these signs or symptoms:
- Weakness of the legs. Lower-extremity weakness or signs of spinal cord damage (eg, loss of sensation at a segmental level, loss of anal sphincter tone and anal wink reflex)
- Fever and back pain
- Pain in your side, lower abdomen or groin
- Abrupt onset or onset during the first few years of life
- Night sweats, cough, and weight loss, especially in a person who has an extensive smoking history
- A mental health disorder
- Blood in your urine
- Red or dark brown urine
- Painful urination
- Difficulty urinating or emptying your bladder
- A strong urge to urinate
- Loss of bladder control
People who have leg weakness should go to the hospital immediately because they may have a spinal cord disorder. People who have fever and back pain should see a doctor within a day because they may have a kidney infection. People who have other warning signs should see a doctor within a day or two. People without warning signs should schedule an appointment as soon as is convenient, usually within a few days to a week, although waiting longer is usually safe if symptoms have been developing over weeks or longer and are mild.
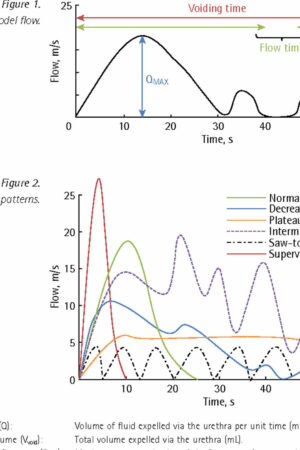
Figure 1. Urinary bladder location
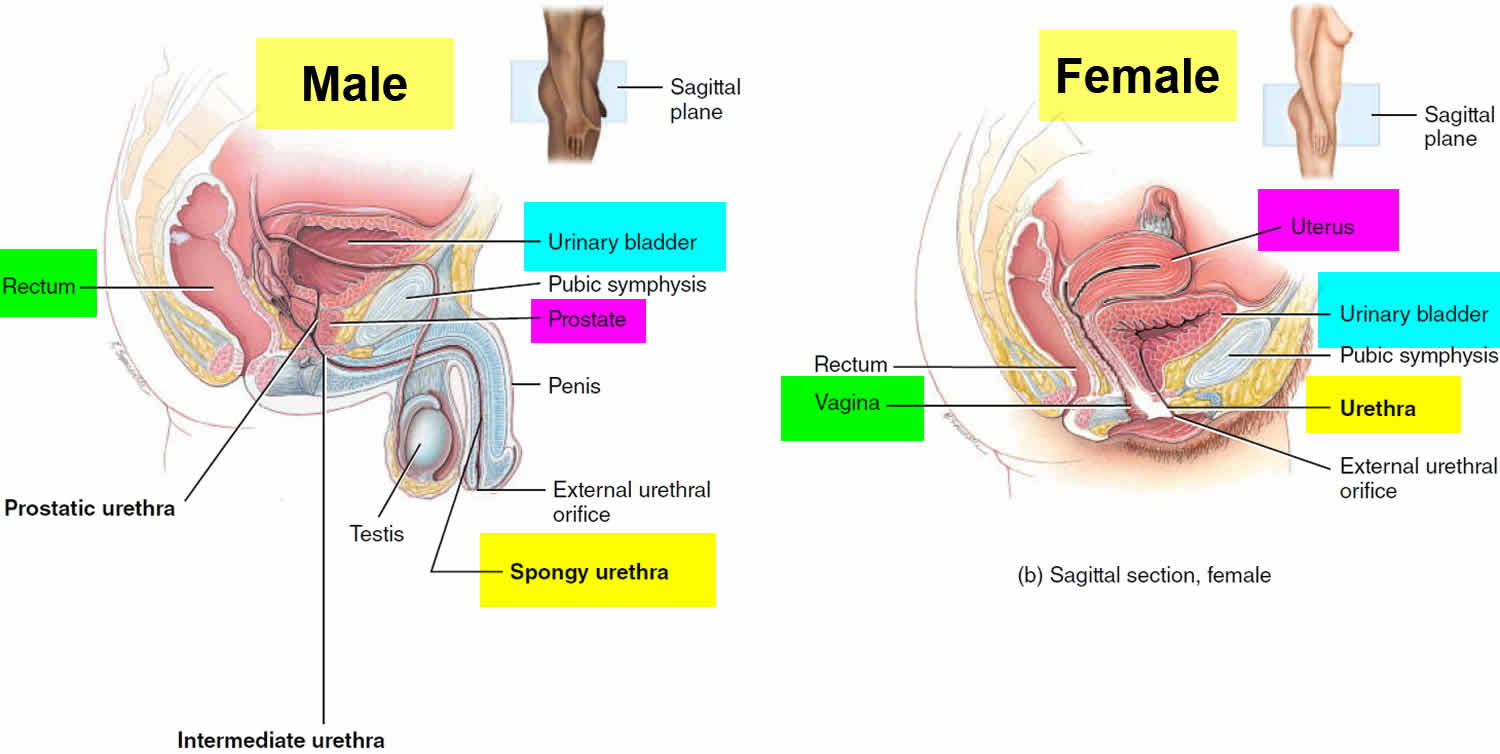
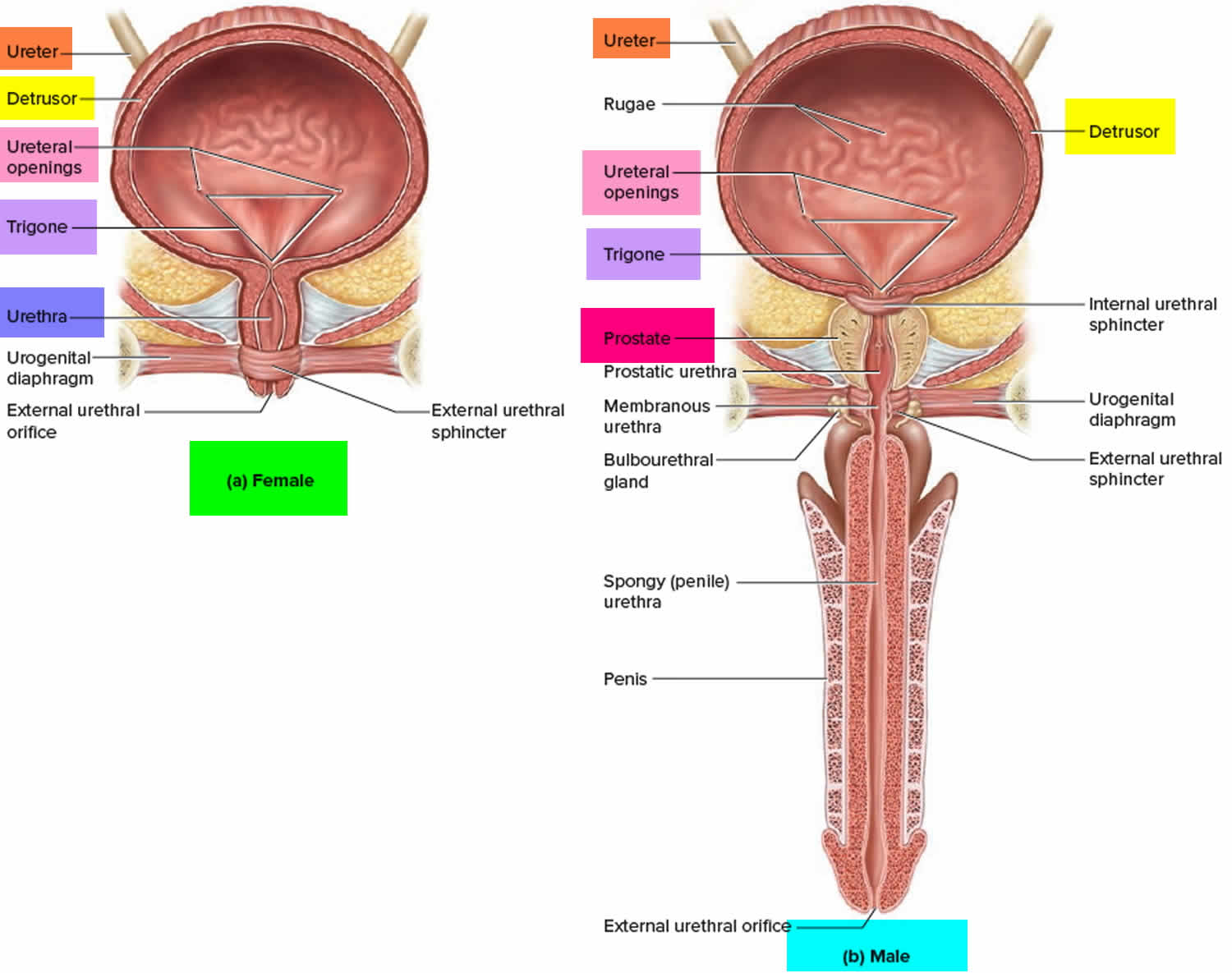
Figure 2. Urinary bladder anatomy
Causes of urinary frequency
Some of the causes of increased urine volume differ from those of too-frequent urination. However, because many people who produce excessive amounts of urine also need to urinate frequently, these two symptoms are often considered together.
The most common causes of urinary frequency are:
- Bladder infections (the most common cause in women and children)
- Urinary incontinence
- Noncancerous enlargement of the prostate gland (benign prostatic hyperplasia—the most common cause in men over 50). Benign prostatic hyperplasia (BPH) is nonmalignant adenomatous overgrowth of the periurethral prostate gland. Symptoms are those of bladder outlet obstruction—weak stream, hesitancy, urinary frequency, urgency, nocturia, incomplete emptying, terminal dribbling, overflow or urge incontinence, and complete urinary retention. Diagnosis is based primarily on digital rectal examination and symptoms; cystoscopy, transrectal ultrasonography, urodynamics, or other imaging studies may also be needed. Treatment options include 5 alpha-reductase inhibitors, alpha-blockers, tadalafil, and surgery.
- Urinary tract infections (UTIs). Urinary tract infections can be divided into upper tract infections, which involve the kidneys (pyelonephritis), and lower tract infections, which involve the bladder (cystitis), urethra (urethritis), and prostate (prostatitis). However, in practice, and particularly in children, differentiating between the sites may be difficult or impossible. Moreover, infection often spreads from one area to the other. Although urethritis and prostatitis are infections that involve the urinary tract, the term urinary tract infection (UTI) usually refers to pyelonephritis and cystitis. Most cystitis and pyelonephritis are caused by bacteria. The most common nonbacterial pathogens are fungi (usually candidal species), and, less commonly, mycobacteria, viruses, and parasites. Nonbacterial pathogens usually affect patients who are immunocompromised; have diabetes, obstruction, or structural urinary tract abnormalities; or have had recent urinary tract instrumentation. Other than adenoviruses (implicated in hemorrhagic cystitis), viruses have no major contribution to UTI in immunocompetent patients. The predominant parasitic causes of urinary tract infections are filariasis, trichomoniasis, leishmaniasis, malaria, and schistosomiasis. Of the parasitic diseases, only trichomoniasis is common in the US, usually as a sexually transmitted disease (STD). Urethritis is usually caused by an STD. Prostatitis is usually caused by a bacterium and is sometimes caused by an STD.
- Stones in the urinary tract. Urinary calculi are solid particles in the urinary system. They may cause pain, nausea, vomiting, hematuria, and, possibly, chills and fever due to secondary infection. Diagnosis is based on urinalysis and radiologic imaging, usually noncontrast helical CT. Treatment is with analgesics, antibiotics for infection, medical expulsive therapy, and, sometimes, shock wave lithotripsy or endoscopic procedures.
The most common causes of polyuria in both adults and children are:
- Uncontrolled diabetes mellitus (most common)
- Drinking too much fluid (polydipsia)
- Diabetes insipidus
- Taking diuretic drugs or substances (which increase the excretion of urine), such as alcohol or caffeine
Diabetes insipidus causes polyuria because of problems with a hormone called antidiuretic hormone (or vasopressin). Antidiuretic hormone helps the kidneys reabsorb fluid. If too little antidiuretic hormone is produced (a condition called central diabetes insipidus) or if the kidneys are unable to properly respond to it (nephrogenic diabetes insipidus), the person urinates excessively.
People with certain kidney disorders (such as interstitial nephritis or kidney damage resulting from sickle cell anemia) may also urinate excessively because these disorders also decrease the amount of fluid reabsorbed by the kidneys.
Table 1. Some causes of urinary frequency
| Cause | Suggestive Findings | Diagnostic Approach |
|---|---|---|
| Benign prostatic hyperplasia or prostate cancer | Progressive onset of urinary hesitancy, incontinence, poor urine stream, a sensation of incomplete voiding | Rectal examination Ultrasonography Cystometry |
| Cystocele | Urinary incontinence Sensation of vaginal fullness Pain or urinary leakage during sexual intercourse |
Pelvic examination Voiding cystourethrography |
| Drugs and substances: Caffeine Alcohol Diuretics |
Urinary frequency in an otherwise healthy patient | Empiric elimination of offending substance (to confirm that frequency resolves) |
| Pregnancy | 3rd trimester of pregnancy | Clinical evaluation |
| Prostatitis | Urgency, dysuria, nocturia, purulent urethral discharge with fever, chills, low back pain, myalgia, arthralgia, and perineal fullness Prostate tender to palpation |
Rectal examination Culture of secretions after prostatic massage |
| Radiation cystitis | History of radiation therapy of the lower abdomen, prostate, or perineum for treatment of cancer | Clinical evaluation Cystoscopy and biopsy |
| Reactive arthritis | Asymmetric arthritis of knees, ankles, and metatarsophalangeal joints Unilateral or bilateral conjunctivitis Small, painless ulcers on the mouth, tongue, glans penis, palms, and soles 1–2 weeks after sexual contact |
STD testing |
| Spinal cord injury or lesion | Lower-extremity weakness, decreased anal sphincter tone, absent anal wink reflex Loss of sensation at a segmental level Injury usually clinically obvious |
MRI of the spine |
| Urethral stricture | Hesitancy, tenesmus, reduced caliber and force of the urine stream | Urethrography |
| Urinary incontinence | Unintentional passage of urine, particularly when bending, coughing, or sneezing | Cystometry |
| Urinary tract calculi | Colicky flank or groin pain | Urinalysis for hematuria Ultrasonography or CT of the kidneys, ureters, and bladder |
| Urinary tract infections | Dysuria and foul-smelling urine, sometimes fever, confusion, and flank pain, particularly in women and girls Dysuria and frequency in young sexually active men (which suggests an STD) |
Urinalysis and culture STD testing |
| Bladder detrusor overactivity | Nocturia, urge incontinence, weak urinary stream, and sometimes urinary retention | Cystometry |
Abbreviation: STD = sexually transmitted disease
Urinary incontinence in children
Urinary incontinence is when a child can’t control her bladder and wets herself during the day. It’s accidental, and there are a few different causes. Urinary incontinence is defined as involuntary voiding of urine ≥ 2 times/month during the day or night. Revised terminology for the time of incontinence has been suggested 2):
- For urinary incontinence during the day: Diurnal incontinence or diurnal wetting
- For urinary incontinence at night: Enuresis or bed-wetting
Diurnal (daytime) incontinence is usually not diagnosed until age 5 or 6. Nocturnal (nighttime) incontinence (that is, enuresis) is usually not diagnosed until age 7. Before this time, enuresis is typically referred to as nighttime wetting 3). These age limits are based on children who are developing typically and so may not be applicable to children with developmental delay. Both nocturnal and diurnal incontinence are symptoms—not diagnoses—and necessitate consideration of an underlying cause.
The age at which children attain urinary continence varies, but > 90% are continent during the day by age 5. Nighttime continence takes longer to achieve. Enuresis affects about 30% of children at age 4, 10% at age 7, 3% at age 12, and 1% at age 18. About 0.5% of adults continue to have nocturnal wetting episodes. Enuresis is more common among boys and when there is a family history 4).
In primary incontinence, children have never achieved urinary continence for ≥ 6 months. In secondary incontinence, children have developed incontinence after a period of at least 6 months of urinary control. An organic cause is more likely in secondary incontinence. Even when there is no organic cause, appropriate treatment and parental education are essential because of the physical and psychologic impact of urine accidents 5).
Sometimes urinary incontinence happens because children’s bladders, genitals, urinary tracts or urethras haven’t developed properly, which means they don’t work properly.
Some children have overactive bladders, which means their bladders don’t store urine the way they’re supposed to. This can make children suddenly feel like they have to do a wee, so they wet themselves.
Other children have underactive bladders, which means they can’t empty their bladders fully, so wee can dribble out before or after they go to the toilet.
Constipation and urinary tract infections (UTI) can cause temporary daytime wetting for some children.
For some children stress can cause urinary incontinence. For example, stressful life events like starting school, the birth of a new sibling, or parents separating can mean that children can’t focus on going to the toilet by themselves. They might start to accidentally wet themselves as a result.
Children who have daytime wetting often also experience bedwetting at night. Usually treatment will focus on daytime wetting first and then look at night-time wetting.
You should see your doctor for advice about treatment and management if your child is older than 5 years and:
- is experiencing urinary incontinence more than once a month
- has never had a period of dryness.
When you see your doctor, your doctor might start by doing a physical examination of your child’s tummy, lower back and genitals. Your doctor might also test your child’s urine.
Your doctor might refer your child to an incontinence specialist.
Symptoms of urinary incontinence in children
The main symptom of urinary incontinence is your child wetting himself during the day because he can’t control his bladder.
How often a child wets herself during the day varies. It can be:
- continuous, which means the child has an uncontrollable dribble of wee, and has never had a period of dryness
- intermittent, which means the child has periods of dryness during the day.
Whether urinary incontinence is continuous or intermittent depends on what’s causing the incontinence.
Treatment for urinary incontinence in children
There are many different treatment options for urinary incontinence, and the right treatment for your child will depend on what’s causing the incontinence.
Behavior modification
The most common treatment for urinary incontinence is behavior modification, which is also called urotherapy.
This involves giving your child information about how his lower urinary tract works, and also about:
- going to the toilet regularly
- not holding on when he needs to wee
- recognizing the signs of needing to do a wee.
Your child might be asked to keep a diary of how often she goes to the toilet, how long in between wees, and how often she wets herself.
The doctor might also ask you to measure the amount of pee your child produces when he goes to the toilet, and how much your child drinks each day. As part of the treatment, your child will probably be asked to wee at particular times throughout the day.
Medication
In some cases, the doctor might prescribe medication to help treat daytime wetting. The type of medication depends on the cause. If your doctor prescribes medication, it’s a good idea to ask why. You can also ask about side effects.
Other treatment options
Depending on the cause of your child’s urinary incontinence, there are other treatment options. These treatment options are for more complex cases of incontinence, and your doctor will usually refer you to a specialist for advice about these options.
Urinary incontinence in adults
Urinary incontinence is involuntary loss of urine; some experts consider it present only when a patient thinks it is a problem. The disorder is greatly underrecognized and underreported. Many patients do not report the problem to their physician, and many physicians do not ask about incontinence specifically. Incontinence can occur at any age but is more common among older people and women, affecting about 30% of older women and 15% of older men.
Incontinence greatly reduces quality of life by causing embarrassment, stigmatization, isolation, and depression. Many older patients are institutionalized because incontinence is a burden to caregivers. In bedbound patients, urine irritates and macerates skin, contributing to sacral pressure ulcer formation. Older people with urgency are at increased risk of falls and fractures.
Types of urinary incontinence in adults
Incontinence may manifest as near-constant dribbling or as intermittent voiding with or without awareness of the need to void. Some patients have extreme urgency (irrepressible need to void) with little or no warning and may be unable to inhibit voiding until reaching a bathroom. Incontinence may occur or worsen with maneuvers that increase intra-abdominal pressure. Postvoid dribbling is extremely common and probably a normal variant in men. Identifying the clinical pattern is sometimes useful, but causes often overlap and much of treatment is the same.
- Urge incontinence. Urge incontinence is uncontrolled urine leakage (of moderate to large volume) that occurs immediately after an urgent, irrepressible need to void. Nocturia and nocturnal incontinence are common. Urge incontinence is the most common type of incontinence in older people but may affect younger people. It is often precipitated by use of a diuretic and is exacerbated by inability to quickly reach a bathroom. In women, atrophic vaginitis, common with aging, contributes to thinning and irritation of the urethra and urgency.
- Stress incontinence. Stress incontinence is urine leakage due to abrupt increases in intra-abdominal pressure (eg, with coughing, sneezing, laughing, bending, or lifting). Leakage volume is usually low to moderate. It is the 2nd most common type of incontinence in women, largely because of complications of childbirth and development of atrophic urethritis. Men can develop stress incontinence after procedures such as radical prostatectomy. Stress incontinence is typically more severe in obese people because of pressure from abdominal contents on the top of the bladder.
- Overflow incontinence. Overflow incontinence is dribbling of urine from an overly full bladder. Volume is usually small, but leaks may be constant, resulting in large total losses. Overflow incontinence is the 2nd most common type of incontinence in men.
- Functional incontinence. Functional incontinence is urine loss due to cognitive or physical impairments (eg, due to dementia or stroke) or environmental barriers that interfere with control of voiding. For example, the patient may not recognize the need to void, may not know where the toilet is, or may not be able to walk to a remotely located toilet. Neural pathways and urinary tract mechanisms that maintain continence may be normal.
- Mixed incontinence. Mixed incontinence is any combination of the above types. The most common combinations are urge with stress incontinence and urge or stress with functional incontinence.
Causes of urinary incontinence in adults
The disorder tends to differ among age groups. With aging, bladder capacity decreases, ability to inhibit urination declines, involuntary bladder contractions (detrusor overactivity) occur more often, and bladder contractility is impaired. Thus, voiding becomes more difficult to postpone and tends to be incomplete. Postvoid residual volume increases, probably to ≤ 100 mL (normal < 50 mL). Endopelvic fascia weakens.
In postmenopausal women, decreased estrogen levels lead to atrophic urethritis and atrophic vaginitis and to decreasing urethral resistance, length, and maximum closure pressure.
In men, prostate size increases, partially obstructing the urethra and leading to incomplete bladder emptying and strain on the detrusor muscle. These changes occur in many normal, continent older people and may facilitate incontinence but do not cause it.
In younger patients, incontinence often begins suddenly, may cause little leakage, and usually resolves quickly with little or no treatment. Often, incontinence has one cause in younger patients but has several in older people.
Transient incontinence
There are several causes of transient incontinence see Table 2.
Table 2. Causes of Transient Incontinence
| Cause | Comments |
|---|---|
| Gastrointestinal disorders | |
| Fecal impaction | Mechanism may involve mechanical disturbance of the bladder or urethra. Patients usually present with urge or overflow incontinence, typically with fecal incontinence. |
| Genitourinary disorders | |
| Atrophic urethritis Atrophic vaginitis |
Thinning of urethral and vaginal epithelium and submucosa may cause local irritation and decrease urethral resistance, length, and maximum closure pressure with loss of the mucosal seal. These disorders are usually characterized by urgency and occasionally by scalding dysuria. |
| Urinary calculi Foreign bodies |
Bladder irritation precipitates spasm. |
| Urinary tract infections | Only symptomatic UTIs cause incontinence. Dysuria and urgency can prevent patients from reaching the toilet before voiding. |
| Neuropsychiatric disorders | |
| Delirium Depression Psychosis |
Awareness of the need or ability to void is impaired. |
| Restricted mobility | |
| Weakness Injury Use of physical restraints |
Access to toilet is impaired. |
| Systemic disorders | |
| Excess urine output due to various disorders (eg, diabetes insipidus, diabetes mellitus) | Frequency, urgency, and nocturia can result. |
| Drugs | |
| Alcohol | Alcohol has a diuretic effect and can cause sedation, delirium, or immobility, which can result in functional incontinence. |
| Caffeine (eg, in coffee, tea, cola and some other soft drinks, cocoa, chocolate, and energy drinks) | Urine production and output are increased, causing polyuria, frequency, urgency, and nocturia. |
| Alpha-adrenergic antagonists (eg, alfuzosin, doxazosin, prazosin, tamsulosin, terazosin) | Bladder neck muscle in women or prostate smooth muscle in men is lax, sometimes causing stress incontinence. |
| Anticholinergics (eg, antihistamines, antipsychotics, benztropine, tricyclic antidepressants) | Bladder contractility can be impaired, sometimes causing urinary retention and overflow incontinence. These drugs also can cause delirium, constipation, and fecal impaction. |
| Calcium channel blockers (eg, diltiazem, nifedipine, verapamil) | Detrusor contractility is decreased, sometimes causing urinary retention and overflow incontinence, nocturia due to peripheral edema, constipation, and fecal impaction. |
| Diuretics (eg, bumetanide, furosemide, [not thiazides]) | Urine production and output are increased, causing polyuria, frequency, urgency, and nocturia. |
| Hormone therapy (systemic estrogen/progestin therapy) | Collagen in the paraurethral connective tissues is degraded, causing ineffective urethral closure. |
| Misoprostol | Misoprostol relaxes the urethra and thus may cause stress incontinence. |
| Psychoactive drugs (eg, antipsychotics, benzodiazepines, sedative-hypnotics, tricyclic antidepressants) | Awareness of the need to void is blunted, and dexterity and mobility are decreased. These drugs can precipitate delirium. |
Established incontinence
Established incontinence is caused by a persistent problem affecting nerves or muscles. Mechanisms usually used to describe these problems are bladder outlet incompetence or obstruction, detrusor overactivity or underactivity, detrusor-sphincter dyssynergia, or a combination. However, these mechanisms are also involved in some transient causes.
- Outlet incompetence. Outlet incompetence is a common cause of stress incontinence. In women, it is usually due to weakness of the pelvic floor or of the endopelvic fascia. Such weakness commonly results from multiple vaginal deliveries, pelvic surgery (including hysterectomy), age-related changes (including atrophic urethritis), or a combination. As a result, the vesicourethral junction descends, the bladder neck and urethra become hypermobile, and pressure in the urethra falls below that of the bladder. In men, a common cause is damage to the sphincter or to the bladder neck and posterior urethra after radical prostatectomy.
- Outlet obstruction. Outlet obstruction is a common cause of incontinence in men, although most men with obstruction are not incontinent. Obstruction in men commonly results from benign prostatic hyperplasia, prostate cancer, or urethral stricture. In both sexes, fecal impaction can cause obstruction. In women, outlet obstruction can result from previous surgery for incontinence or from a prolapsed cystocele that causes the urethra to kink during straining to void. Obstruction leads to a chronically overdistended bladder, which loses its ability to contract; then the bladder does not empty completely, resulting in overflow. Obstruction also may lead to detrusor overactivity and urge incontinence. If the detrusor muscle loses its ability to contract, overflow incontinence may follow. Some causes of outlet obstruction (eg, large bladder diverticula, cystoceles, bladder infections, calculi, and tumors) are reversible.
- Detrusor overactivity. Detrusor overactivity is a common cause of urge incontinence in older and younger patients. The detrusor muscle contracts intermittently for no apparent reason, usually when the bladder is partially or nearly full. Detrusor overactivity may be idiopathic or may result from dysfunction of the frontal micturition inhibitory center (commonly due to age-related changes, dementia, or stroke) or outlet obstruction. Detrusor overactivity (hyperactivity) with impaired contractility is a variant of urge incontinence characterized by urgency, frequency, a weak flow rate, urinary retention, bladder trabeculation, and a postvoid residual volume of > 50 mL. This variant may mimic prostatism in men or stress incontinence in women.
- Overactive bladder. Overactive bladder is a term sometimes used to describe urinary urgency (with or without incontinence) that is often accompanied by urinary frequency and nocturia.
- Detrusor underactivity. Detrusor underactivity causes urinary retention and overflow incontinence in about 5% of patients with incontinence. It may be caused by injury to the spinal cord or to nerve roots supplying the bladder (eg, by disk compression, tumor, or surgery), by peripheral or autonomic neuropathies, or by other neurologic disorders (see table Causes of Established Incontinence). Anticholinergics and opioids greatly decrease detrusor contractility; these drugs are common transient causes. The detrusor may become underactive in men with chronic outlet obstruction as the detrusor is replaced by fibrosis and connective tissue, preventing the bladder from emptying even when the obstruction is removed. In women, detrusor underactivity is usually idiopathic. Less severe detrusor weakness is common among older women. Such weakness does not cause incontinence but can complicate treatment if other causes of incontinence coexist.
- Detrusor-sphincter dyssynergia. Detrusor-sphincter dyssynergia (loss of coordination between bladder contraction and external urinary sphincter relaxation) may cause outlet obstruction, with resultant overflow incontinence. Dyssynergia is often due to a spinal cord lesion that interrupts pathways to the pontine micturition center, which coordinates sphincter relaxation and bladder contraction. Rather than relaxing when the bladder contracts, the sphincter contracts, obstructing the bladder outlet. Dyssynergia causes severe trabeculation, diverticula, a “Christmas tree” deformation of the bladder seen on cystogram, hydronephrosis, and renal failure.
- Functional impairment. Functional impairment (eg, cognitive impairment, reduced mobility, reduced manual dexterity, coexisting disorders, lack of motivation), particularly in older patients, may contribute to established incontinence but rarely causes it.
Table 3. Causes of established incontinence
| Urodynamic Diagnosis | Some Neurologic Causes | Some Nonneurologic Causes |
|---|---|---|
| Bladder outlet incompetence | Lower motor neuron lesion (rare). In men, radical prostatectomy* | Intrinsic sphincter deficiency Urethral hypermobility In women, multiple vaginal deliveries, pelvic surgery (eg, hysterectomy), age-related changes (eg, atrophic urethritis) In men, prostate surgery |
| Bladder outlet obstruction | Spinal cord lesion causing detrusor-sphincter dyssynergia (rare) | Anterior urethral stricture Urethral diverticula (rarely) or large bladder diverticula (very rarely) Bladder calculi Bladder neck suspension surgery In women, cystocele (if large) In men, benign prostatic hyperplasia or prostate cancer |
| Detrusor overactivity | Alzheimer disease Spinal cord injury/dysfunction Multiple sclerosis Stroke |
Bladder carcinoma Cystitis Idiopathic Outlet obstruction or incompetence |
| Detrusor underactivity | Autonomic neuropathy (eg, due to diabetes, alcoholism, or vitamin B12 deficiency) Disk compression Plexopathy Spinal neural tube defect (less often, may cause overactivity) Surgical damage (eg, anteroposterior resection) Tumor |
Chronic bladder outlet obstruction. Idiopathic (common among women) |
| Detrusor-sphincter dyssynergia | Spinal cord lesion Brain lesion affecting pathways to the pontine micturition center |
Voiding dysfunction of childhood (poor relaxation of the sphincter with bladder contraction can result from the fear of bed wetting or soiling of clothes) |
Footnote: * Other prostate surgical procedures rarely cause established incontinence.
Urinary frequency diagnosis
Many people are embarrassed to discuss problems related to urination with their doctor. But because some disorders that cause excessive urination are quite serious, people who urinate excessively should be evaluated by a doctor. The following information can help people know when to see a doctor and what to expect during the evaluation.
Doctors first ask questions about the person’s symptoms and medical history and then do a physical examination. What they find during the history and physical examination often suggests a cause of excessive urination and the tests that may need to be done.
Medical history
Doctors ask about:
- Amounts of fluid drunk and urinated to determine whether the problem is related to urinary frequency or to polyuria
- If urinary frequency is present, patients are asked about acuity of onset, presence or absence of irritative symptoms (eg, irritation, urgency, dysuria), obstructive symptoms (eg, hesitancy, poor flow, sensation of incomplete voiding, nocturia), and recent sexual contacts.
- How long symptoms have been present
- Whether any other urination problems are present
- Whether the person is taking diuretics (drugs and other substances that increase urine production), including beverages that contain caffeine
- Review of systems should cover symptoms suggestive of a cause, including fever, flank or groin pain, and hematuria (infection); missed menses, breast swelling, and morning sickness (pregnancy); and arthritis and conjunctivitis (reactive arthritis).
- Past medical history should ask about known causes, including prostate disease and previous pelvic radiation therapy or surgeries. Drugs and diet are reviewed for the use of agents that increase urine output (eg, diuretics, alcohol, caffeinated beverages).
Some obvious findings may give clues to the cause of frequent urination. Pain or burning during urination (dysuria), fever, and back or side pain may indicate an urinary tract infection (UTI) or calculi. In a person who drinks large amounts of beverages that contain caffeine or who has just started treatment with a diuretic, the diuretic substance is a likely cause. A man who has other problems with urination, such as difficulty starting urination, a weak urine stream, and dribbling at the end of urination, may have a prostate disorder.
Prior pelvic surgery suggests incontinence. Weak urine stream, nocturia, or both suggests benign prostatic hyperplasia (BPH). Urinary frequency in an otherwise healthy young patient may be due to excessive intake of alcohol or caffeinated beverages. Gross hematuria (blood in urine) suggests UTI and calculi in younger patients and genitourinary cancer in older patients.
Some obvious findings may also give clues to the cause of polyuria. For example, polyuria that starts during the first few years of life is likely caused by an inherited disorder such as central or nephrogenic diabetes insipidus or type 1 diabetes mellitus.
Physical examination
Examination focuses on the genitourinary system.
Any urethral discharge or any lesions consistent with sexually transmitted diseases are noted. Rectal examination in men should note the size and consistency of the prostate and rectal tone; pelvic examination in women should note the presence of any cystocele. Patients should be instructed to cough while the urethra is observed for signs of urinary leakage.
The costovertebral angle should be palpated for tenderness, and the abdominal examination should note the presence of any masses or suprapubic tenderness.
Neurologic examination should test for lower-extremity weakness and loss of sensation.
In women, the physical examination usually includes a pelvic examination and the taking of samples of cervical and vaginal fluid to check for sexually transmitted diseases. In men, the penis is examined for presence of a discharge, and doctors do a digital rectal examination to examine the prostate.
Testing
All patients require urinalysis and culture, which are easily done and can detect infection and hematuria.
The need for other testing depends on what doctors find during the history and physical examination.
If doctors are not sure whether the person is actually producing more urine than normal, they may collect and measure the amount of urine produced over 24 hours. If people actually have polyuria, doctors measure the blood glucose level. If diabetes mellitus is not the cause of polyuria and no other cause, such as excess intravenous fluids, is clearly responsible, other testing is necessary. The levels of electrolytes and concentration of certain salts (osmolarity) are measured in the blood, urine, or both, often after the person is deprived of water for a time and after the person is given antidiuretic hormone.
Cytoscopy, cystometry, and urethrography can be done to diagnose cystitis, bladder outlet obstruction, and cystocele. Prostate-specific antigen level determination, ultrasonography, and prostate biopsy may be required, especially in older men, to differentiate benign prostatic hyperplasia (noncancerous enlargement of the prostate gland) from prostate cancer.
Urinary frequency treatment
Urinary frequency treatment involves treating the underlying cause or condition.
References [ + ]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}