



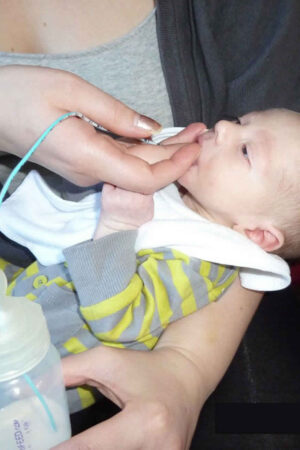

Thumb sucking
Thumb sucking is one of the most common habits of children 1). A habit is a behavior that your child does over and over again, almost without thinking. Thumb or finger sucking starts early in life, with 90 percent of newborns showing some form of hand sucking by two hours of age 2). Sucking is one of the most common reflexes seen in infants. It manifests when they are in womb around 29 wk of age 3). This is the first pattern of behavior observed in infant. Infants and young children may use finger, thumb, pacifiers, or other objects to feel secure and learn the outside world. This is commonly seen when the child is anxious, insecure or surrounded by strangers and in families when they are separated from their parents. Sucking habit induces sleep and hence makes infant and child calm and relaxed 4).
Hand sucking is naturally developed in 89% of infants in the second month and increases by first year of life 5). It is normal up to 2–4 years of age. Thumb sucking is normal in infants and young children and should cause no permanent problems if it is not continued past the age of 5. Likewise, it is generally harmless for infants to use pacifiers.
The prevalence of oral habit has been reported up to 88% and 30% in high school girls and boys, respectively 6). 34% of prevalence has been reported in other literature 7). It has been documented that parental education, child’s nutrition, and sucking habits are associated with each other 8). However, higher prevalence rate has been claimed with high stress level among children in recent time 9).
After second year of age, sucking habit should start decreasing and should appear only when child goes to sleep 10). When it continues in mixed and permanent dentition, it becomes a parental concern, as it is believed to affect growth of maxilla palate leading to skeletal changes, showing side effects on developing occlusion and resulting in malocclusion and constriction of skeletal structures 11).
The American Academy of Pediatric Dentistry states that most children stop thumb sucking on their own between the ages of 2 and 4. The Academy states there is no reason to be concerned until the front teeth start erupting. At this point, some problems may occur, including bite problems, or protruding front teeth. It may result in anterior open bite, increased over jet, lingual inclination of lower incisor and labial inclination of maxillary anterior, posterior crossbite, deep palate, compensatory tongue thrust, and sometimes speech defect 12). The changes in dentition depend upon duration and frequency of habit being performed. During active eruption phase of permanent teeth, children who perform sucking habit for longer duration (more than 6 hrs in a day), especially during sleep, tend to develop minor skeletal abnormalities and sever dentoalveolar changes 13).
Other problems that may occur with thumb sucking are sore thumbs, infections, and calluses on the thumb.
It is thought that pacifier use may actually be better than thumb sucking for the following reasons:
- Pacifiers are softer and cause less damage to the teeth.
- The plastic rim on the pacifier provides some relief of the tension placed on the teeth.
- Pacifiers can be cleaned.
The use of pacifier may cause severe and harmful effects on dentition if used for more than 5-year-old child 14). Consult your child’s dentist if you are concerned with your child’s thumb sucking. Generally, it is not a problem for children under the age of five.
Why do habits start?
Habits can be comforting for children. Sucking is a good example. As toddlers leave behind the baby stage, habits like thumb-sucking can be a way of soothing stress or anxiety.
Sometimes habits happen because children are bored. That is, the behavior is just how children entertain themselves. For example, children are actually more likely to bite their nails while watching TV or doing nothing at all than when they’re feeling anxious.
Sometimes habits start for practical reasons but keep going when the practical reasons have gone. For example, young children with colds often pick their noses to clear them. Children who keep picking even after they’ve learned to blow their noses probably have habits.
You’re a role model for your child. If you see your child starting a habit, perhaps ask yourself whether it’s one of your own habits. For example, nail-biting might be passed on within a family.
Note: some toddlers seem to get comfort from some common but slightly unusual behavior, including body-rocking, head-rolling and head-banging. Most children stop this behaviour by the time they’re five years old.
When should children quit thumb sucking?
Until the age of five, thumb suckers shouldn’t change the shape of their jaw. Their baby teeth may be out of line a little but the greatest concern is the jaw which will require orthodontic work to treat the alignment problem.
After the age of five their jaw shape is more like to be impacted and the teeth are more likely be misaligned. It’s a good idea to try getting your child out of the habit once they have their fifth birthday.
Does thumb sucking cause orthodontic problems?
Yes, in some children, thumb and finger sucking past the age of five can cause problems such as:
- Teeth alignment: The most obvious sign that a child sucks their thumb is changes to the front teeth. The sucked thumb or finger can cause the upper front teeth to protrude forward. The constant pressure from the hand in their mouth can also cause the lower front teeth to tip forward.
- Jaw: While not as common as the changes to teeth, thumb sucking can change the shape of a child’s jaw. The upper jaw can narrow so it doesn’t match the bottom jaw. A cross-bite may occus because the upper and lower teeth don’t fit together. An open bite can also occur with thumb sucking. The upper teeth don’t overlap the bottom teeth when the back teeth are together. The thumb or finger has created an opening between the top and bottom front teeth and prevents them from meeting.
- Speech: A child’s speech can be impacted by bucked teeth or an open bite. Straight front teeth help pronounce some letters. A lisp may be heard when they say the s and z sounds. Without speech therapy and orthodontic treatment later on, the lisp can remain into adulthood.
- Face shape: The shape of your jaw influences the shape of your face. An overbite where the front teeth are pushed forward to accommodate the thumb, is likely to change the overall look and shape of a child’s face. Orthodontic treatment can reverse the face shape changes.
Factors that influence the severity of the problem
There are three main factors that determine the likelihood of thumb and finger sucking causing orthodontic problems.
- Duration. If the thumb sucking habit doesn’t last long or the child does not suck their thumb for long on a given day, then it’s unlikely they will do any damage to their teeth or jaw.
- Frequency. Some children will only suck their thumb for a short time during the day while other children will suck their thumb all day and even night while they sleep. It’s when the frequency is high that children are most at risk of doing damage.
- Type of sucking. Not all thumb sucking is the same. Some children just let their finger sit in their mouth but others use more force. The harder a child sucks on their finger, the more damage they can do.
Dummies vs Thumb Sucking
Parents will often wonder if they should have introduced their baby to a dummy rather than let them suck on their thumb, however a dummy isn’t much better. They can still cause jaw and teeth alignment problems if they use it long and often enough.
Research shows that there can be significant dental arch and occlusion characteristics in dummy users between 24 and 36 months compared to children who stopped using one by the age of 12 months. However, significant problems occur in long-term users after the age of five but ideally the dummy should be discarded before 3 years of age. No shape or brand of dummy is better than another at reducing teeth and jaw risks.
How to stop thumb sucking?
Childhood habits like thumb or fingers sucking are not usually something to worry about. In fact, many young kids suck their thumbs, probably as a way to cope with stress or anxiety. Thumb sucking can be soothing and comforting, and can make a child feel safe.
Most childhood habits go away on their own — especially if a parent ignores them. But if kids continue to suck their thumbs into the school-age years, it may cause shifts in their teeth and affect the way the mouth grows.
When child performs sucking habit in the first year of life, parents can move away his/her thumb smoothly and attract the child to other things.
Some tips for breaking habits
- Gently remind your child about the habit. For example, if your child sucks on a finger, you can say, ‘Please don’t chew on your finger – it’s a bit yucky’. Remind positively rather than threaten them with punishment for thumb sucking.
- Try to encourage your child to do something else during idle times. For example, you could encourage your child to play with a toy that has moveable parts while watching television.
- Keep them busy. If they are doing things with their hands, particularly if they are dirty, they are less likely to put their hands in their mouth.
- Try to find out why your child is doing the habit, and suggest an alternative.
- Habits can come in pairs, like sucking a thumb and pulling hair. When you stop the thumb-sucking, the hair-pulling might also stop.
- Praise will go a long way towards stopping habits. For example, you can say, ‘That’s great. I can hear your words clearly when your fingers aren’t in your mouth’.
- If your child doesn’t seem to be outgrowing her/his thumb sucking, try some positive reinforcement. Reward her/his with an extra bedtime story or other favorite treat when she doesn’t suck her thumb. Be generous with the praise, too. It can also help eliminate anything causing her anxiety or stress. And remember, most habits in young kids are just passing phases.
- Wearing a glove can remind a child not to put their hand in their mouth.
- Applying nail polish can act as a reminder and encouragement not to damage their manicure by putting their thumb in their mouth.
- A sour tasting liquid on their fingers to make them less appealing
- For older kids, an orthodontic plate can make it less enticing to put their thumb in if the space has been taken up by the plate.
Habits in children with additional needs
Children with additional needs might have more habits than typically developing children, or habits that are more pronounced. A psychologist or other specialist experienced with additional needs can help if you’re looking for more information.
When to get help for habits
At about three years of age, thumb-sucking and finger-sucking can become a problem for children’s teeth development. If your child is still finger-sucking beyond three years, talk to your pharmacist about using other approaches, like a sticking plaster or a paint-on solution. The solution makes fingers taste yucky.
If you’re concerned that your child’s sucking is causing problems, you could see your dentist about using a palate barrier. This device makes it uncomfortable for children to suck thumbs or fingers.
If you think anxiety might be the reason behind a habit, you might need to deal with the cause of the anxiety. Talk to your doctor about getting a referral to another health professional. For example, a psychologist can teach your child some simple steps to stop the habit.
Management of sucking habit depends upon the age. Counseling by pediatric dentist and pediatrician is important. Historically, correction of thumb or finger habit revolves around direct counseling of child, encouragement to improve self-confidence by rewarding the child, appliance therapy, and, in case of more complex dental changes, orthodontic therapy along with habit breaking appliances 15). Appliance therapy involves use of either fixed or removable design in form of palatal crib or spurs. It is reminder therapy for child to make the habit unpleasant and difficult to practice. However, it causes difficulty in speech and eating and can cause iatrogenically inflicted wound and make child emotionally disturbed 16).
Tongue, palatal cribs, spurs, palatal bars, hay rakes, cage type appliances and other sharp points employ an aversive negative stimulus to cease the undesirable oral habits. They are moderately effective and may trigger unexpected behavior sometimes. However, emotional disturbances, difficulty in speech and eating, and iantrogenically self-inflicted wounds can occur with such appliances. Hay rake and cage type appliances tend to get mutilated or destroyed while eating or due to habitual sucking habit. It reminds the child as punitive therapy to cease the habit 17). In 1991, Haskell and Mink introduced Blue grass appliance, also known as habit correction roller which gained universal attention and acceptance 18). It is user friendly, nondestructive, easy to wear appliance replacing the common destructive habits. It is useful in avoiding traditional physical barriers of appliance in form of cribs and helping child with positive reinforcement. Later, similar appliance called Lingual Pearl was used as a habit breaking and for multiple clinical applications 19). Further, Baker modified blue grass appliance with multiple rollers/beads and thus expanding its use from primary to permanent dentition 20).
Figure 1. Blue grass appliance for finger sucking
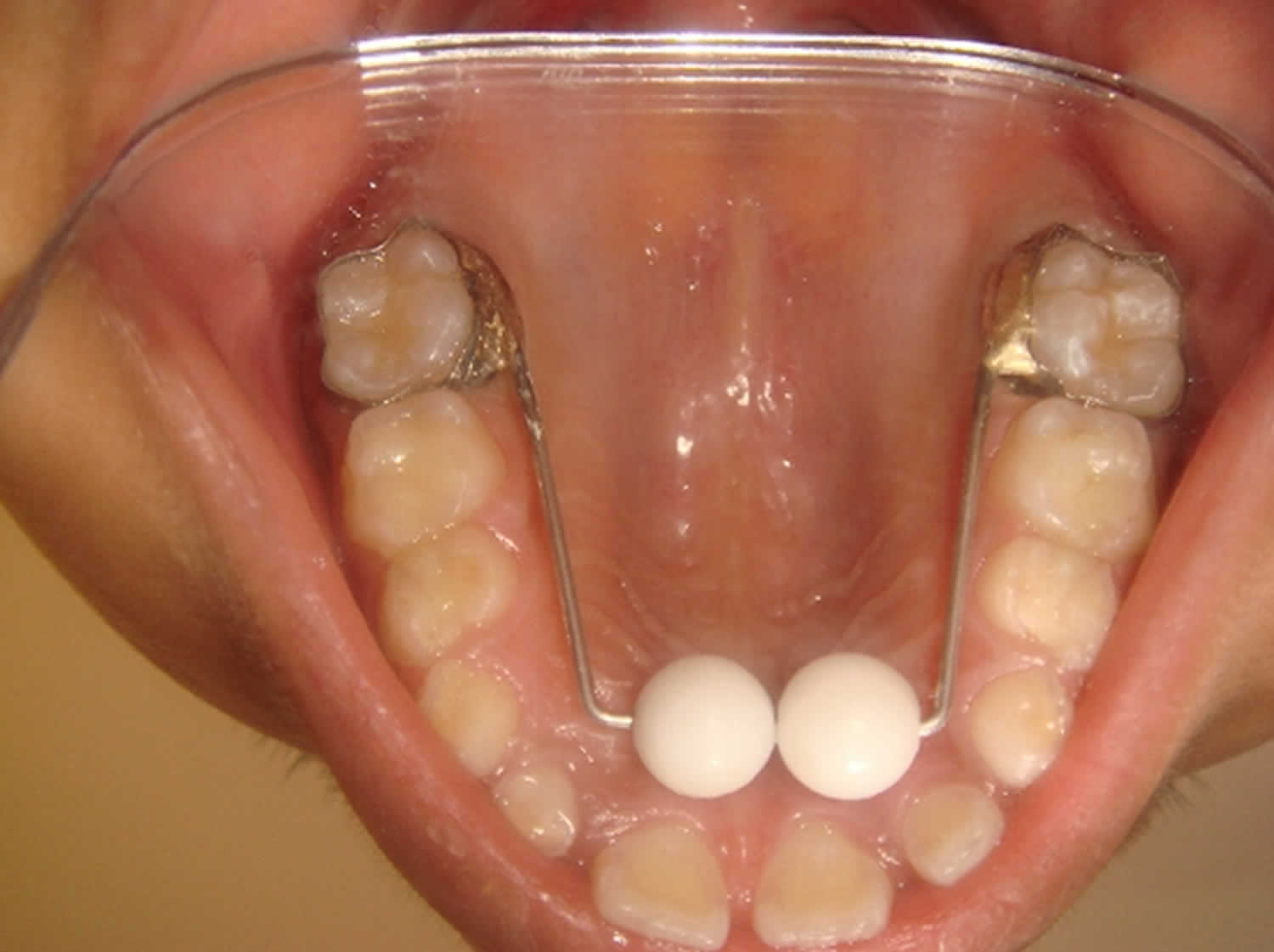
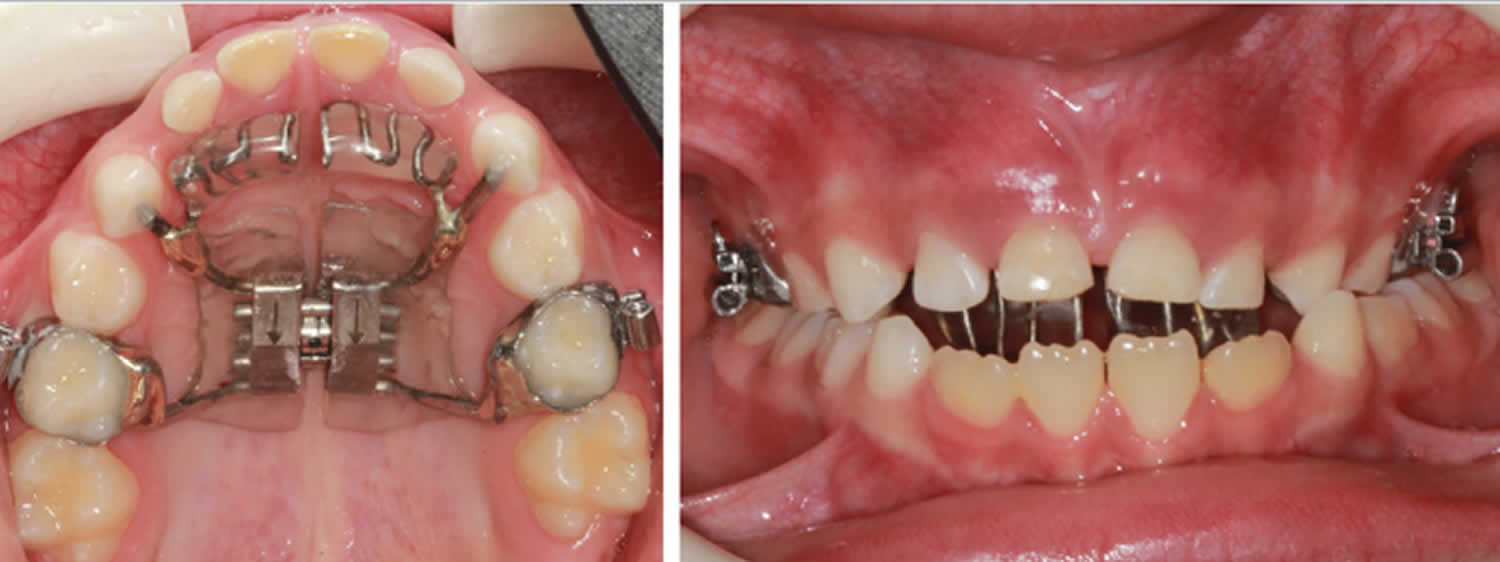
Figure 2. Palatal expander

Figure 3. Haas expander with tongue crib
References [ + ]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}