Contents
Beta thalassemia
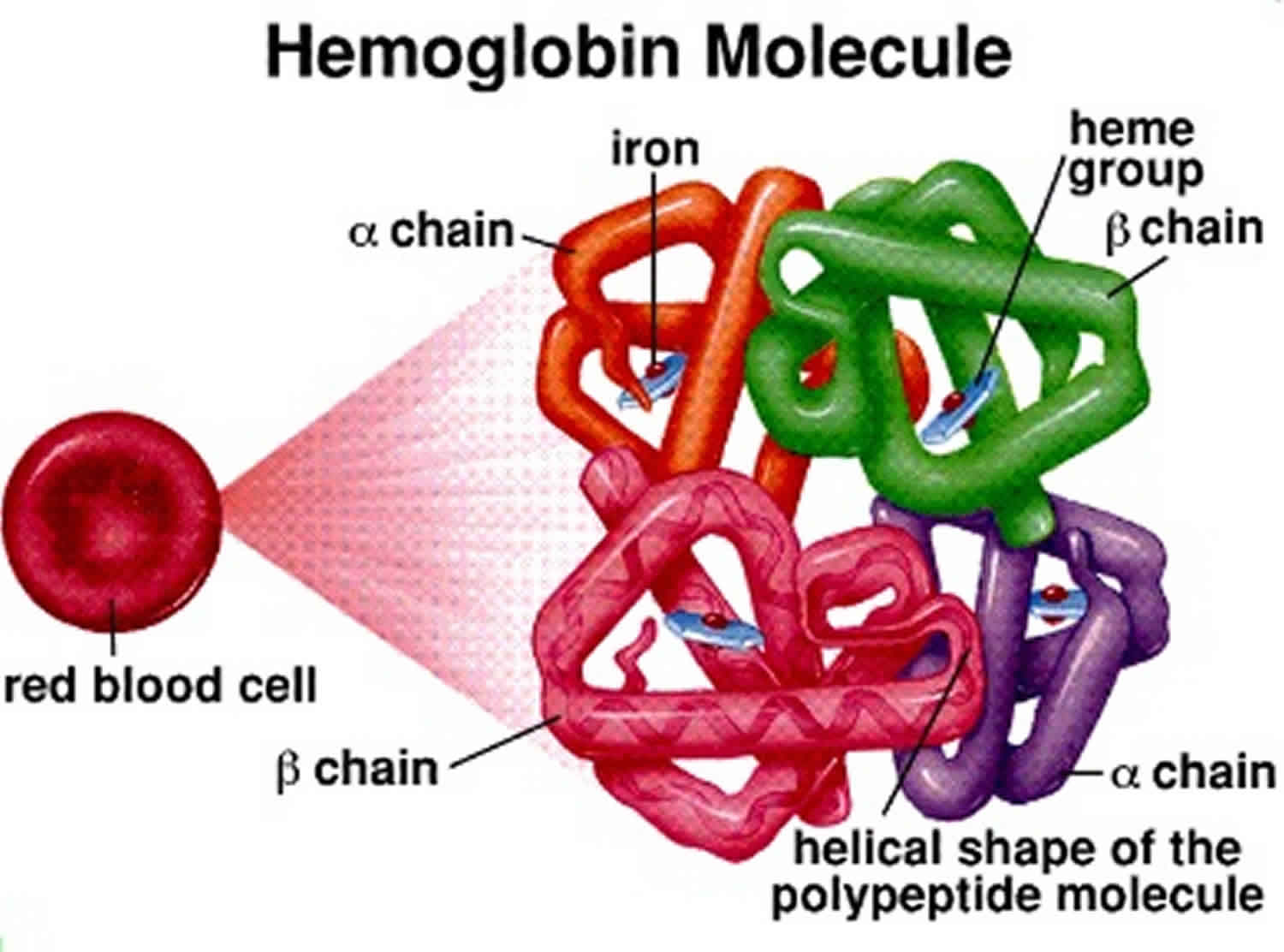
Beta thalassemia (β-thalassemia) also called Mediterranean anemia or erythroblastic anemia, is a blood disorder that reduces the production of hemoglobin (reduced synthesis of the hemoglobin subunit beta chains). Hemoglobin is the iron-containing protein in red blood cells that carries oxygen to cells throughout the body and low levels of hemoglobin lead to a lack of oxygen in many parts of the body. People with beta thalassemia have anemia (shortage of red blood cells), which can cause paleness, weakness, fatigue, and more serious complications. People with beta thalassemia are at an increased risk of developing abnormal blood clots.
Beta thalassemia is a fairly common blood disorder worldwide. Thousands of infants with beta thalassemia are born each year. Beta thalassemia occurs most frequently in people from Mediterranean countries, North Africa, the Middle East, India, Central Asia, and Southeast Asia.
Beta-thalassemia is characterized by microcytic hypochromic anemia, an abnormal peripheral blood smear with nucleated red blood cells, and reduced amounts of hemoglobin A (HbA) on hemoglobin analysis.
Beta thalassemia is classified into two types depending on the severity of symptoms: thalassemia major also known as Cooley’s anemia and thalassemia intermedia. Of the two types, thalassemia major is more severe.
The signs and symptoms of thalassemia major appear within the first 2 years of life. Children develop life-threatening anemia. They do not gain weight and grow at the expected rate (failure to thrive) and may develop yellowing of the skin and whites of the eyes (jaundice). Affected individuals may have an enlarged spleen, liver, and heart, and their bones may be misshapen. Some adolescents with thalassemia major experience delayed puberty. Many people with thalassemia major have such severe symptoms that they need frequent blood transfusions to replenish their red blood cell supply. Over time, an influx of iron-containing hemoglobin from chronic blood transfusions can lead to a buildup of iron in the body, resulting in liver, heart, and hormone problems.
Thalassemia intermedia is milder than thalassemia major. The signs and symptoms of thalassemia intermedia appear in early childhood or later in life. Affected individuals have mild to moderate anemia and may also have slow growth and bone abnormalities.
Can an individual with beta-thalassemia minor donate blood?
When an individual chooses to donate blood, he/she is typically examined and asked specific questions about his/her medical history (to make sure that donating blood isn’t unsafe for the individual donating or for the recipient). During this process, the individual’s hematocrit value (or hemoglobin level) is tested to make sure that the individual does not have anemia and is not likely to become anemic after donation. In order to donate blood, an individual’s hemoglobin level must be at a specific level, which is established by the U.S. Food and Drug Administration (FDA). Usually, individuals with hemoglobin levels that are too low are temporarily not permitted to donate blood. A low hematocrit level is one of the most common reason people are temporarily disqualified or “deferred” from donating blood, but some donors can actually have anemia and still be eligible to donate.
People who have beta-thalassemia minor and are interested in donating blood should speak with their healthcare provider.
My partner and I have both been diagnosed with beta-thalassemia minor. What is the likelihood of having a child without beta-thalassemia?
Thalassemia major and thalassemia intermedia are inherited in an autosomal recessive pattern, which means both copies of the HBB gene in each cell have mutations (see Figure 1 below). The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene and are referred to as carriers, but they typically do not show signs and symptoms of the condition. When two carriers have children, each child has a 25% (1 in 4) chance to be affected, a 50% (1 in 2) chance to be a carrier like each parent, and a 25% (1 in 4) chance to be unaffected and not a carrier. Sometimes, however, people (carriers) with only one HBB gene mutation in each cell develop mild anemia. These mildly affected people are said to have ‘beta-thalassemia minor’ or ‘beta-thalassemia trait’ 1).
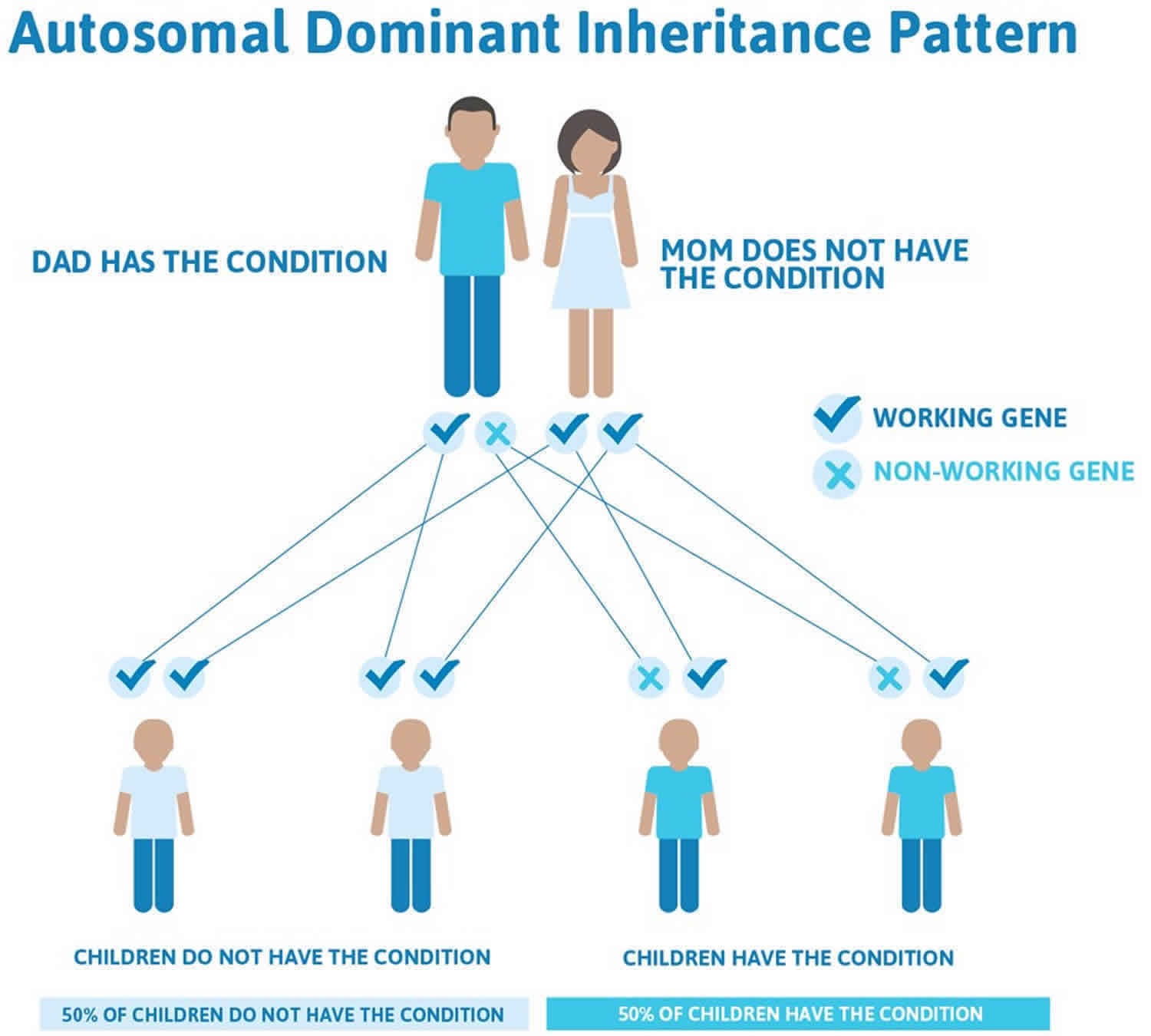
In a small percentage of families, the HBB gene mutation is inherited in an autosomal dominant manner (see Figure 2 below). In these cases, one copy of the altered gene in each cell is sufficient to cause the signs and symptoms of beta thalassemia 2).
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Beta thalassemia types
There are three types of beta thalassemia, depending upon whether one or two beta globin genes are mutated, and the severity of symptoms 3):
- Beta thalassemia minor, or beta thalassemia trait, happens when one of the beta globin genes is mutated. People with this condition typically have very mild symptoms and require no treatment, but they can pass thalassemia on to their children. Usually, they are mildly anemic and their red blood cells are smaller than normal.
- Thalassemia major also called Cooley’s anemia – the more severe form, causing severe anemia and enlarged liver and spleen (hepatosplenomegaly). This form usually becomes apparent before 2 years of age. If not treated, it causes failure to thrive and a shortened life expectancy. Treatment involves regular transfusions and chelation therapy to reduce iron overload. Treatment allows for normal growth and development. Bone marrow transplantation or cord blood transplantation may eliminate the need for regular treatment.
- Thalassemia intermedia – the less severe form, becoming apparent later and causing milder anemia that does not require regular blood transfusions. People with this form are also at risk for iron overload.
- Dominant beta thalassemia is an extremely rare form in which individuals who have one mutated HBB gene develop certain symptoms associated with beta thalassemia. Affected individuals may develop mild to moderate anemia, jaundice, and an abnormally enlarged spleen (splenomegaly).
The signs and symptoms of thalassemia major appear within the first 2 years of life. Children develop life-threatening anemia. They do not gain weight and grow at the expected rate (failure to thrive) and may develop yellowing of the skin and whites of the eyes (jaundice). Affected individuals may have an enlarged spleen, liver, and heart, and their bones may be misshapen. Some adolescents with thalassemia major experience delayed puberty. Many people with thalassemia major have such severe symptoms that they need frequent blood transfusions to replenish their red blood cell supply. Over time, an influx of iron-containing hemoglobin from chronic blood transfusions can lead to a buildup of iron in the body, resulting in liver, heart, and hormone problems.
Thalassemia intermedia is milder than thalassemia major. The signs and symptoms of thalassemia intermedia appear in early childhood or later in life. Affected individuals have mild to moderate anemia and may also have slow growth and bone abnormalities.
Beta thalassemia causes
Mutations in the HBB gene cause beta thalassemia. The HBB gene provides instructions for making a protein called beta-globin. In extremely rare cases, a loss of genetic material (deletion) that includes the HBB gene causes beta thalassemia. Beta-globin is a component (subunit) of hemoglobin. Hemoglobin consists of four protein subunits, typically two subunits of beta-globin and two subunits of another protein called alpha-globin.
To date, more than 250 mutations that could cause beta thalassemia have been reported all over the world 4).
Some mutations in the HBB gene prevent the production of any beta-globin. The absence of beta-globin is referred to as beta-zero (B0) thalassemia. Other HBB gene mutations allow some beta-globin to be produced but in reduced amounts. A reduced amount of beta-globin is called beta-plus (B+) thalassemia. Having either B0 or B+ thalassemia does not necessarily predict disease severity, however; people with both types have been diagnosed with thalassemia major and thalassemia intermedia.
A lack of beta-globin leads to a reduced amount of functional hemoglobin. Without sufficient hemoglobin, red blood cells do not develop normally, causing a shortage of mature red blood cells. The low number of mature red blood cells leads to anemia and other associated health problems in people with beta thalassemia.
Beta thalassemia inheritance pattern
Thalassemia major and thalassemia intermedia are inherited in an autosomal recessive pattern, which means both copies of the HBB gene in each cell have mutations (see Figure 1). The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene and are referred to as carriers, but they typically do not show signs and symptoms of the condition. When two carriers have children, each child has a 25% (1 in 4) chance to be affected, a 50% (1 in 2) chance to be a carrier like each parent, and a 25% (1 in 4) chance to be unaffected and not a carrier. Sometimes, however, people (carriers) with only one HBB gene mutation in each cell develop mild anemia. These mildly affected people are said to have ‘beta-thalassemia minor’ or ‘beta-thalassemia trait’ 5).
In a small percentage of families, the HBB gene mutation is inherited in an autosomal dominant manner (Figure 2). In these cases, one copy of the altered gene in each cell is sufficient to cause the signs and symptoms of beta thalassemia 6).
Figure 1. Beta thalassemia autosomal recessive inheritance pattern
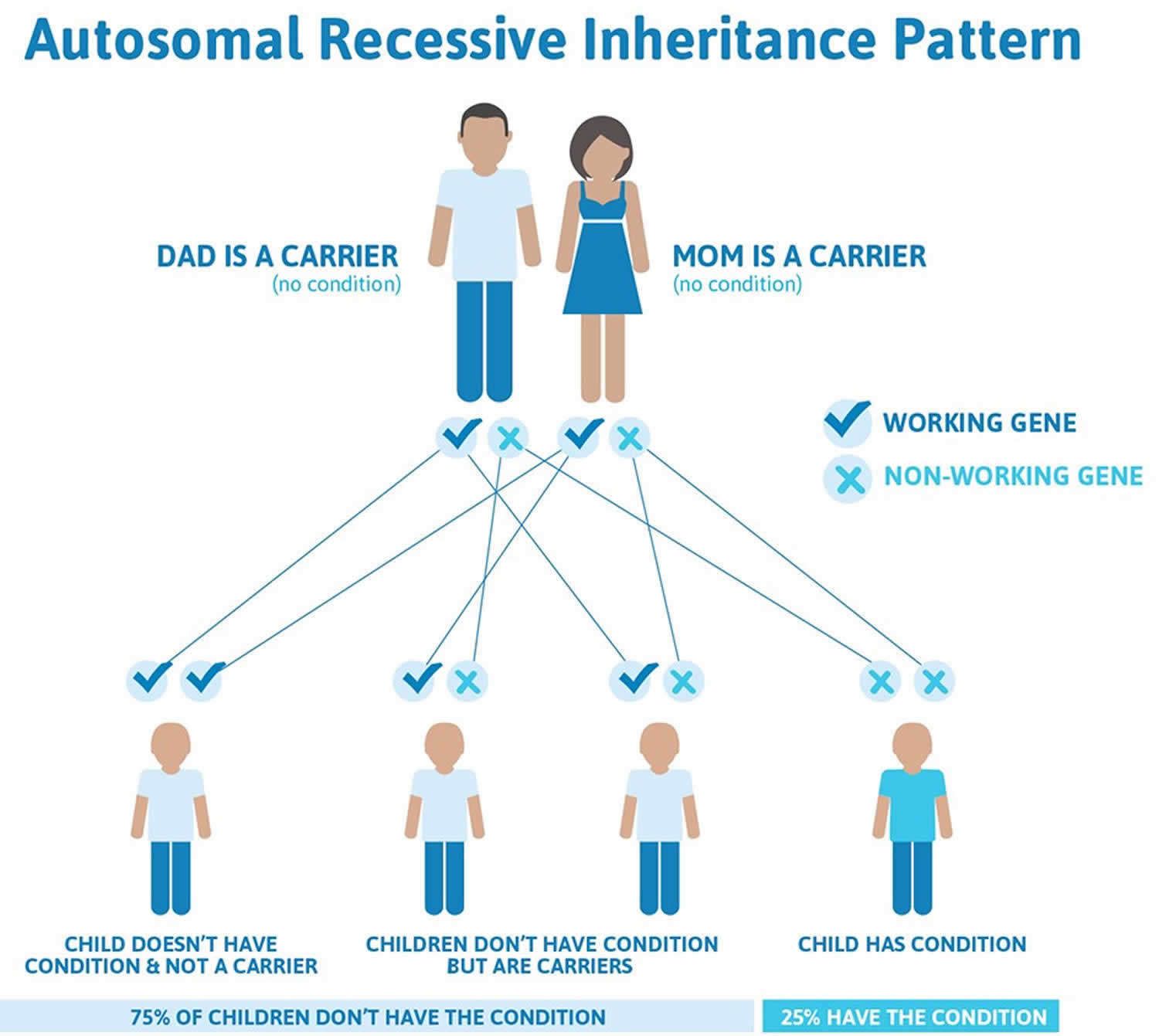
Figure 2. Beta thalassemia autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Beta thalassemia symptoms
The symptoms and severity of beta thalassemia varies greatly from one person to another. Individuals with beta thalassemia minor do not develop symptoms of the disorder but may have a mild anemia. Many individuals with beta thalassemia minor go through life never knowing they carry an altered gene for the disorder.
A beta thalassemia major diagnosis is usually made during the first two years of life and individuals require regular blood transfusions and lifelong medical care to survive. When the disorder develops later during life, a diagnosis of beta thalassemia intermedia is given; individuals may only require blood transfusions on rare, specific instances.
Beta thalassemia major (Cooley’s anemia)
Beta thalassemia major also known as Cooley’s anemia, is the most severe form of beta thalassemia. Affected infants exhibit symptoms within the first two years of life, often between 3 and 6 months after birth. The full or classic “description” of beta thalassemia major tends to primarily occur in developing countries. Most individuals will not develop the severe symptoms discussed below. Although beta thalassemia major is a chronic, lifelong illness, if individuals follow the current recommended treatments, most individuals can live happy, fulfilling lives.
Severe anemia develops and is associated with fatigue, weakness, shortness of breath, dizziness, headaches, and yellowing of the skin, mucous membranes and whites of the eyes (jaundice). Affected infants often fail to grow and gain weight as expected based upon age and gender (failure to thrive). Some infants become progressively pale (pallor). Feeding problems, diarrhea, irritability or fussiness, recurrent fevers, abnormal enlargement of the liver (hepatomegaly), and the abnormal enlargement of the spleen (splenomegaly) may also occur.
Splenomegaly may cause abdominal enlargement or swelling. Splenomegaly may be associated with an overactive spleen (hypersplenism), a condition that can develops because too many blood cells build up and are destroyed within the spleen. Hypersplenism can contribute to anemia in individuals with beta thalassemia and cause low levels of white blood cells, increasing the risk of infection, and low levels of platelets, which can lead to prolonged bleeding.
If untreated, additional complications can develop. Beta thalassemia major can cause the bone marrow, the spongy material within certain bones, to expand. Bone marrow is where most of the blood cells are produced in the body. The bone marrow expands because it is trying to compensate for chronic anemia. This abnormal expansion causes bones to become thinner, wider and brittle. Affected bones may grow abnormally (bone deformities), particularly the long bones of the arms and legs and certain bones of the face. When facial bones are affected it can result in distinctive facial features including an abnormally prominent forehead (frontal bossing), full cheek bones (prominent malar eminence), a depressed bridge of the nose, and overgrowth (hypertrophy) of the upper jaw (maxillae), exposing the upper teeth. The affected bones have an increased fracture risk, particularly the long bones of the arms and legs. Some individuals may develop ‘knock knees’ (genu valgum), a condition in which the legs bend inward so that when a person is standing the knees will touch even if the ankles and feet are not.
Even when treated, complications may develop, specifically the buildup of iron in the body (iron overload). Iron overload results from the blood transfusions required to treat individuals with beta thalassemia major. In addition, affected individuals experience greater iron absorption from the gastrointestinal tract, which contributes to iron overload (although this primarily occurs in untreated individuals). Iron overload can cause tissue damage and impaired function of affected organs such as the heart, liver and endocrine glands. Iron overload can damage the heart causing abnormal heart rhythms, inflammation of the membrane (pericardium) that lines the heart (pericarditis), enlargement of the heart and disease of the heart muscle (dilated cardiomyopathy). Heart involvement can progress to life-threatening complications such as heart failure. Liver involvement can cause scarring and inflammation of the liver (cirrhosis) and high pressure of the main liver vein (portal hypertension). Endocrine gland involvement can cause insufficiency of certain glands such as the thyroid (hypothyroidism) and, in rare cases, diabetes mellitus. Iron overload can also be associated with growth retardation and the failure or delay of sexual maturation.
Additional symptoms that may occur include masses that form because of blood cell production outside of the bone marrow (extramedullary hematopoiesis). These masses primarily form in the spleen, liver, lymph nodes, chest, and spine and can potentially cause compression of nearby structures and a variety of symptoms. Affected individuals may develop leg ulcers, an increased risk of developing blood clots within a vein (venous thrombosis) and decreased bone mineralization resulting in brittle bones that are prone to fracture (osteoporosis).
Beta thalassemia intermedia
Individuals diagnosed with beta thalassemia intermedia have a widely varied expression of the disorder. Moderately severe anemia is common and affected individuals may require periodic blood transfusions. Each individual case is unique. Common symptoms include pallor, jaundice, leg ulcers, gallstones (cholelithiasis), and abnormal enlargement of the liver and spleen. Moderate to severe skeletal malformations (as described in beta thalassemia major) may also occur.
Dominant beta thalassemia
Dominant beta thalassemia is an extremely rare form in which individuals who have one mutated HBB gene develop certain symptoms associated with beta thalassemia. Affected individuals may develop mild to moderate anemia, jaundice, and an abnormally enlarged spleen (splenomegaly).
Beta thalassemia complications
Complications associated with beta thalassemia, aside from anemia, are as follows:
- Extramedullary hematopoiesis
- Medical complications from long-term transfusional therapy – Iron overload and transfusion-associated infections (eg, hepatitis); iron overload cardiomyopathy accounts for the majority of deaths in thalassemia patients 7).
- Increased risk for infections resulting from asplenia (eg, encapsulated organisms such as pneumococcus) or from iron overload (eg, Yersinia species)
- Cholelithiasis (eg, bilirubin stones)
- Excess iron. Kids who have beta thalassemia can end up with too much iron in their bodies, either from the disease itself or from getting repeated blood transfusions. Excess iron can cause damage to the heart, liver, and endocrine system.
- Bone deformities and broken bones. Beta thalassemia can cause bone marrow to expand, making bones wider, thinner, and more brittle. This makes bones more likely to break and can lead to abnormal bone structure, particularly in the bones of the face and skull.
- Enlarged spleen. The spleen helps fight off infections and filters out unwanted materials, such as dead or damaged blood cells, from the body. Beta thalassemia can cause red blood cells to die off at a faster rate, making the spleen work harder, which makes it grow larger. A large spleen can make anemia worse and may need to be removed if it gets too big.
- Infections. Children with beta thalassemia have a higher risk of infection, especially if they’ve had their spleens removed.
- Slower growth rates. The anemia resulting from beta thalassemia can cause children to grow more slowly and also can lead to delayed puberty.
Beta thalassemia diagnosis
In most cases, beta thalassemia is diagnosed before a child’s second birthday. Children with beta thalassemia major may have a swollen abdomen or symptoms of anemia or failure to thrive.
If the doctor suspects beta thalassemia, he or she will take a blood sample for testing. Blood tests can reveal red blood cells that are pale, varied in shape and size, and smaller than normal. They also can detect low red blood cell counts and cells with an uneven distribution of hemoglobin, which causes them to look like a bull’s-eye when seen through a microscope.
Blood tests also can measure the amount of iron in the blood. Usually the diagnosis is confirmed by a blood test called a hemoglobin electrophoresis and/or a DNA test for abnormal hemoglobin genes.
Molecular genetic testing can confirm a beta thalassemia diagnosis. Molecular genetic testing can detect mutations in the HBB gene known to cause the disorder, but is available only as a diagnostic service at specialized laboratories. Molecular genetic testing is not necessary to make a diagnosis of beta thalassemia and is generally used to identify at-risk, asymptomatic relatives, to aid prenatal diagnosis, and to attempt to predict the progression or severity of the disease in specific cases.
If both parents are carriers of the beta thalassemia disorder, doctors can conduct tests on a fetus before birth. This is done through either:
- chorionic vilius sampling, which takes place about 11 weeks into pregnancy and involves removing a tiny piece of the placenta for testing
- amniocentesis, which is usually done about 16 weeks into the pregnancy and involves removing a sample of the fluid that surrounds the fetus
If one parent carries a beta thalassemia gene and the other carries a different gene that also affects beta globin, such as a sickle gene, their child could have a significant blood disorder (such as a form of sickle cell disease called sickle-beta thalassemia). Therefore, people who carry beta thalassemia genes should seek genetic counseling if they’re considering having children so they can understand the risks.
Beta thalassemia treatment
Individuals with beta thalassemia major and intermedia will benefit from referral to a thalassemia treatment center. These specialized centers provide comprehensive care for individuals with beta thalassemia including the development of specific treatment plans, monitoring and follow up of affected individuals, and state-of-the-art medical care. Treatment at such a center ensures that individuals and their family members will be cared for by a professional healthcare team (physicians, nurses, physical therapists, social workers and genetic counselors) experienced in the treatment of individuals with beta thalassemia. Genetic counseling is recommended for affected individuals and their families. Psychosocial support for the entire family is essential as well.
Specific therapeutic procedures and interventions may vary, depending upon numerous factors, such as the specific type of beta thalassemia; the progression of the disease; the presence or absence of certain symptoms; severity of the disease upon diagnosis; an individual’s age and general health; and/or other elements. Decisions concerning the use of a particular drug regimen and/or other treatments should be made by physicians and other members of the health care team in consultation with the patient based upon the specifics of his or her case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
The amount of treatment that beta thalassemia requires depends on how severe the symptoms are. For most children with beta thalassemia trait, whose only symptom may be mild anemia from time to time, no medical treatment will be necessary. It is important that individuals with beta thalassemia minor be correctly diagnosed, however, in order to avoid unnecessary treatments for similarly-appearing conditions such as iron deficiency anemia.
However, the blood counts in beta thalassemia trait look a lot like the blood counts in iron deficiency anemia, which is a very common disorder. It’s important for doctors to know when children have beta thalassemia trait so that they do not treat them with iron if it’s not needed.
Doctors also might recommend a folic acid supplement for kids with moderate cases of anemia to help boost production of new red blood cells. Supplementation with folic acid, a B vitamin, boosts the production of red blood cells in certain individuals.
Some children with moderate anemia may require an occasional blood transfusion, particularly after surgery. Those with severe cases of beta thalassemia major, on the other hand, may require regular blood transfusions their entire lives to keep them healthy. During blood transfusions, they’re given blood from donors with matching blood types. Over time, this can cause a build-up of iron in the body, so kids who receive frequent blood transfusions may have to take medications to remove excess iron from their bodies.
Some individuals may be treated by the surgical removal of the spleen (splenectomy). An abnormally enlarged spleen (splenomegaly) can cause severe pain and contribute to anemia. Splenomegaly can cause low levels of the blood cells (platelets) that allow the blood to clot. An enlarged spleen in individuals with beta thalassemia may occur due to increased destruction of red blood cells, the formation of blood cells outside of the bone marrow (extramedullary hematopoiesis), repeated blood transfusions, or iron overload. If other forms of therapy fail, removal of the spleen may be considered. Splenectomy has led to improvement in certain symptoms associated with beta thalassemia. However, this surgical procedure carries risks, which are weighed against benefits in each individual case. If a splenectomy is required, one month before the surgery pneumococcal conjugate vaccine should be given. In addition, antibiotic prophylaxis, usually penicillin 250 mg twice a day, is given the first two years after surgery and for children younger than 16 years. Because of advances in the treatment of beta thalassemia in the past several years, splenectomy is rarely necessary as a treatment for affected individuals.
Individuals with beta thalassemia major and intermedia may develop iron overload, which occurs because of two reasons. First, blood transfusions cause the accumulation of excess iron in the body. Second, beta thalassemia can cause increased absorption of dietary iron by the gastrointestinal tract. The body has no normal way to remove excess iron. In individuals who receive regular blood transfusions, iron overload primarily occurs because of treatment. Iron overload causes a variety of symptoms affecting various body organ systems. Iron overload is treated by medications that remove excess iron from the body such as deferoxamine. Deferoxamine is an iron chelator, a drug that binds to iron in the body allowing it to be dissolved in water and excreted from the body through the kidneys. Other oral iron chelators, such as deferiprone and deferasirox, have also been used to lower iron levels.
Treatment of additional complications of beta thalassemia or iron overload is symptomatic and supportive. Special attention is recommended for the early diagnosis and prompt treatment of heart (cardiac) disease potentially associated with iron overload. Cardiac disease is the main life-threatening complication in individuals with beta thalassemia.
Research into treating beta thalassemia with experimental gene therapies is ongoing. But for now, it can only be cured by a procedure called a bone marrow transplant (also called a stem cell transplant). Bone marrow, which is found inside bones, produces blood cells. In a bone marrow transplant, children are first given high doses of radiation or drugs to destroy the defective bone marrow. The bone marrow is then replaced with cells from a compatible donor, usually a healthy sibling or other relative. Bone marrow transplants carry many risks, so they usually are done only in the most severe cases of thalassemia.
If your child has beta thalassemia, support groups are available to help your family cope with the obstacles presented by the disease.
Beta thalassemia prognosis
Individuals with thalassemia minor (thalassemia trait) usually have mild, asymptomatic microcytic anemia. This state does not result in mortality or significant morbidity.
The prognosis of patients with thalassemia major is highly dependent on the patient’s adherence to long-term treatment programs, namely the hypertransfusion program and lifelong iron chelation. Allogeneic bone marrow transplantation may be curative.
Morbidity and mortality
The major causes of morbidity and mortality in beta thalassemia are anemia and iron overload. The severe anemia resulting from this disease, if untreated, can result in high-output cardiac failure; the intramedullary erythroid expansion may result in associated skeletal changes such as cortical bone thinning. The long-term increase in red-cell turnover causes hyperbilirubinemia and bilirubin-containing gallstones.
Increased iron deposition resulting from lifelong transfusions and enhanced iron absorption results in secondary iron overload. This overload causes clinical problems similar to those observed with primary hemochromatosis (eg, endocrine dysfunction, liver dysfunction, cardiac dysfunction).
A broad spectrum of neurological complications has also been reported in beta thalassemia complications, although most were subclinical. These have included the following 8):
- Cognitive impairment
- Abnormal findings on evoked potentials
- Cerebrovascular disease
- Peripheral neuropathy.
References [ + ]




{kind=link}