Contents
Alexander disease
Alexander disease is an extremely rare, usually progressive and fatal, neurological disorder. It is one of a group of disorders, called leukodystrophies (diseases of the white matter of the brain), that involve the destruction of myelin. Myelin is the fatty covering that insulates nerve fibers and promotes the rapid transmission of nerve impulses. If myelin is not properly maintained, the transmission of nerve impulses could be disrupted. As myelin deteriorates in leukodystrophies such as Alexander disease, nervous system functions are impaired.
There is a marked deficit in myelin formation in most early onset patients with Alexander disease, and sometimes in later onset patients, particularly in the front (frontal lobes) of the brain’s two hemispheres (cerebrum). However, white matter defects are sometimes not observed in later onset individuals. Instead, the unifying feature among all Alexander disease patients is the presence of abnormal protein deposits known as “Rosenthal fibers” throughout certain regions of the brain and spinal cord (central nervous system [CNS]). These aggregates occur inside specialized cells called astroglial cells or astrocytes, a common cell type in the CNS that helps support and nourish other cells in the brain and spinal cord (central nervous system). Accordingly, it is more appropriate to consider Alexander disease a disease of astrocytes (an astrogliopathy) than a white matter disease (leukodystrophy).
The prevalence of Alexander disease is unknown. Alexander disease has been estimated to occur at a frequency of about 1 in 1 million births 1). About 500 cases have been reported since the disorder was first described in 1949 2). No racial, ethnic, geographic, or sex preference has been observed, nor is any expected given the de novo (new) nature of the mutations responsible for most cases. Although initially diagnosed primarily in young children, it is now being observed with similar frequency at all ages.
Most cases of Alexander disease begin before age 2 and are described as the infantile form. Signs and symptoms of the infantile form typically include an enlarged brain and head size (megalencephaly), seizures, stiffness in the arms and/or legs (spasticity), intellectual disability, and developmental delay. Less frequently, onset occurs later in childhood (the juvenile form) or in adulthood. Common problems in juvenile and adult forms of Alexander disease include speech abnormalities, swallowing difficulties, seizures, and poor coordination (ataxia). Rarely, a neonatal form of Alexander disease occurs within the first month of life and is associated with severe intellectual disability and developmental delay, a buildup of fluid in the brain (hydrocephalus), and seizures.
No specific therapy is currently available for Alexander disease. Management is supportive and includes attention to general care, physical and occupational therapy, nutritional requirements, antibiotic treatment for any infection, and antiepileptic drugs for seizure control 3).
Physical and occupational therapy and speech therapy may be recommended depending on the specific signs and symptoms present. Physical and occupational therapy may be indicated in people with developmental and language delays.
Figure 1. Alexander disease MRI brain
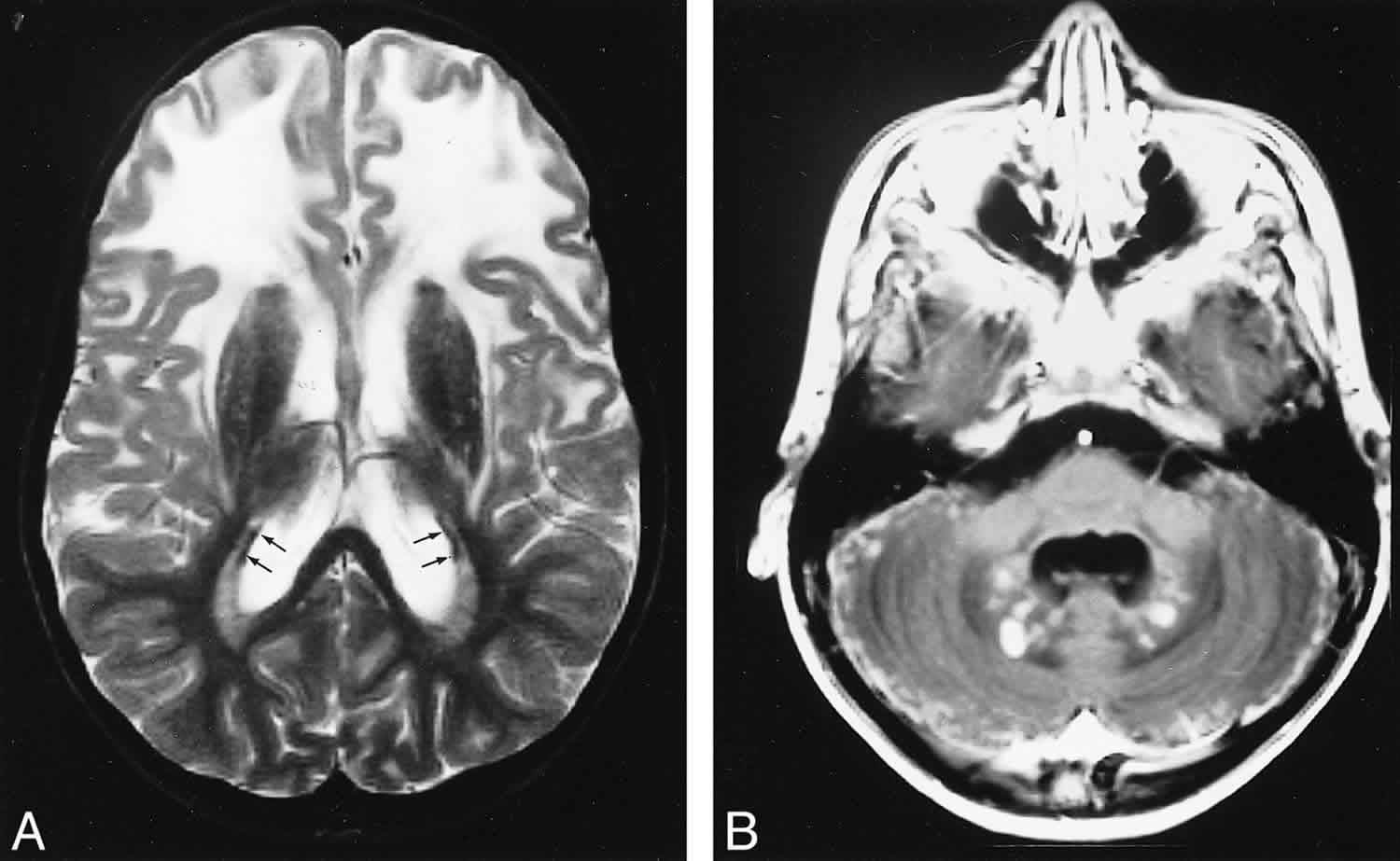
Footnote: Late MR imaging study of a patient with presumed juvenile Alexander disease, obtained at the age of 10 years. A and B, There is extensive white matter involvement with frontal preponderance (A). The basal ganglia are dark and atrophic on T2-weighted images (A). A thin periventricular rim of low signal intensity is just visible (arrows, A). After contrast administration, enhancement of the entire cerebellar surface and dentate nucleus is seen (B).
[Source 4) ]Infantile Alexander disease
Infantile Alexander disease leads to symptoms in the first two years of life; while some children die in the first year of life, a larger number live to be 5-10 years old. The usual course of infantile Alexander disease is progressive, leading to eventual severe mental retardation and spastic quadriparesis (spasms that may involve all four limbs). However, in some children the degree of disability develops slowly over several years, and some children retain responsiveness and emotional contact until near the end of their lives. Feeding often becomes a problem, and assisted feeding (as with a nasogastric tube) may become necessary. Their head circumference is often enlarged. Children with hydrocephalus caused by Alexander disease usually have increased intracranial pressure and a more rapid progression of the disease. Generally, the earlier the age of onset of Alexander disease, the more severe and rapid the course.
Below is a list of the clinical terms of some of the symptoms and pathologies of infantile Alexander disease, along with definitions of each term. Please keep in mind that severity and symptoms will vary, and so all children will not have all symptoms.
- Megalencephaly: Megalencephaly means that the brain is abnormally large; this can be associated with delayed development, convulsive disorders, corticospinal (brain cortex and spinal cord) dysfunction, and seizures.
- Hydrocephaly: Literally means “water on the brain.” Characterized by the accumulation of fluid in the brain or between the brain and the skull. Can cause pressure on the brain, resulting in developmental defects. Also can lead to an abnormally large head size (to greater than 90% of normal).
- Failure to thrive: A general term meaning the the child is not growing and gaining weight at the expected rate.
- Seizures: The brain controls how the body moves by sending electrical signals. Seizures (also called convulsions) occur when the normal signals from the brain are changed. Severity of a seizure can vary dramatically. Some people may only shake slightly and do not lose consciousness. Other people may become unconscious and have violent shaking of the entire body.
- Spasticity/spastic quadriparesis: This means that the child tends to suffer spasms, or involuntary contractions of muscles. Muscles are abnormally stiff and movement is restricted. Quadriparesis means that all four limbs are involved.
- Progressive psychomotor retardation: This can include difficulties with walking, speech difficulties, and mental regression. Eventually this can lead to loss of all meaningful contact with the environment. Progressive means that the condition worsens as time goes on.
Juvenile Alexander disease
Juvenile Alexander disease is characterized by difficulty with talking and swallowing and the inability to cough. There can also be weakness and spasticity of the extremities, particularly the legs. Unlike in the infantile form of the disease, mental ability and head size may be normal. Age of onset is usually between the ages of 4 and 10. Survival can extend several years following onset of symptoms, with occasional longer survival into middle age.
The course of juvenile Alexander disease may involve signs of swallowing or speech difficulty, vomiting, ataxia, and/or spasticity. Kyophoscoliosis can occur. Mental function often slowly declines, although in some cases the intellectual skills remain intact.
Pathologically, whereas the infantile form of Alexander disease generally affects the brain, the juvenile form generally leads to changes in the brain stem rather than in the brain. There are many Rosenthal fibers (as in infantile Alexander Disease), but the lack of myelin is less prominent than in the infantile form.
Adult-onset Alexander disease
Adult-onset Alexander disease is the most rare of the forms, and also is generally the most mild. Onset can be anywhere from the late teens to very late in life. In older patients ataxia (impaired coordination) often occurs and difficulties in speech articulation, swallowing, and sleep disturbances may occur. Symptoms can be similar to those in juvenile disease, although the disease may also be so mild that symptoms are not even noticed until an autopsy reveals the presence of the Rosenthal fibers. Symptoms may resemble multiple sclerosis or a tumor.
Alexander disease cause
About 95% of Alexander disease cases are caused by mutations in a gene called GFAP gene. The cause of the other 5% of cases is not known. The GFAP gene provides instructions for making a protein called glial fibrillary acidic protein that is found exclusively in astrocytes in the central nervous system (brain and spinal cord). Several molecules of glial fibrillary acidic protein bind together to form intermediate filaments, which provide support and strength to cells. Mutations in the GFAP gene lead to the production of a structurally altered glial fibrillary acidic protein. The altered protein is thought to impair the formation of normal intermediate filaments. As a result, the abnormal glial fibrillary acidic protein likely accumulates in astroglial cells, leading to the formation of Rosenthal fibers, which impair cell function. It is not well understood how impaired astroglial cells contribute to the abnormal formation or maintenance of myelin, leading to the signs and symptoms of Alexander disease.
How the GFAP mutations produce Alexander disease is not known. The Rosenthal fibers, which contain GFAP, accumulate throughout the surfaces of the brain (cerebral cortex), in the white matter of the brain, and in the lower regions of the brain (brainstem), and the spinal cord, and primarily appear under the innermost of the protective membranes (meninges) surrounding the brain and spinal cord (pia mater); under the lining of the fluid-filled cavities (ventricles) of the brain (subependymal regions); and around blood vessels (perivascular regions). Studies in mice indicate that the mutations act by producing a new, toxic effect, rather than by interfering with the normal function of GFAP. This toxic effect may be due to the presence of the Rosenthal fibers, or to the very large, abnormal amounts of GFAP that accumulate in Alexander astrocytes, or both. Astrocytes perform many critical functions in the CNS, and several of these are affected by the GFAP mutations, but the importance of these changes to the disease is not yet known.
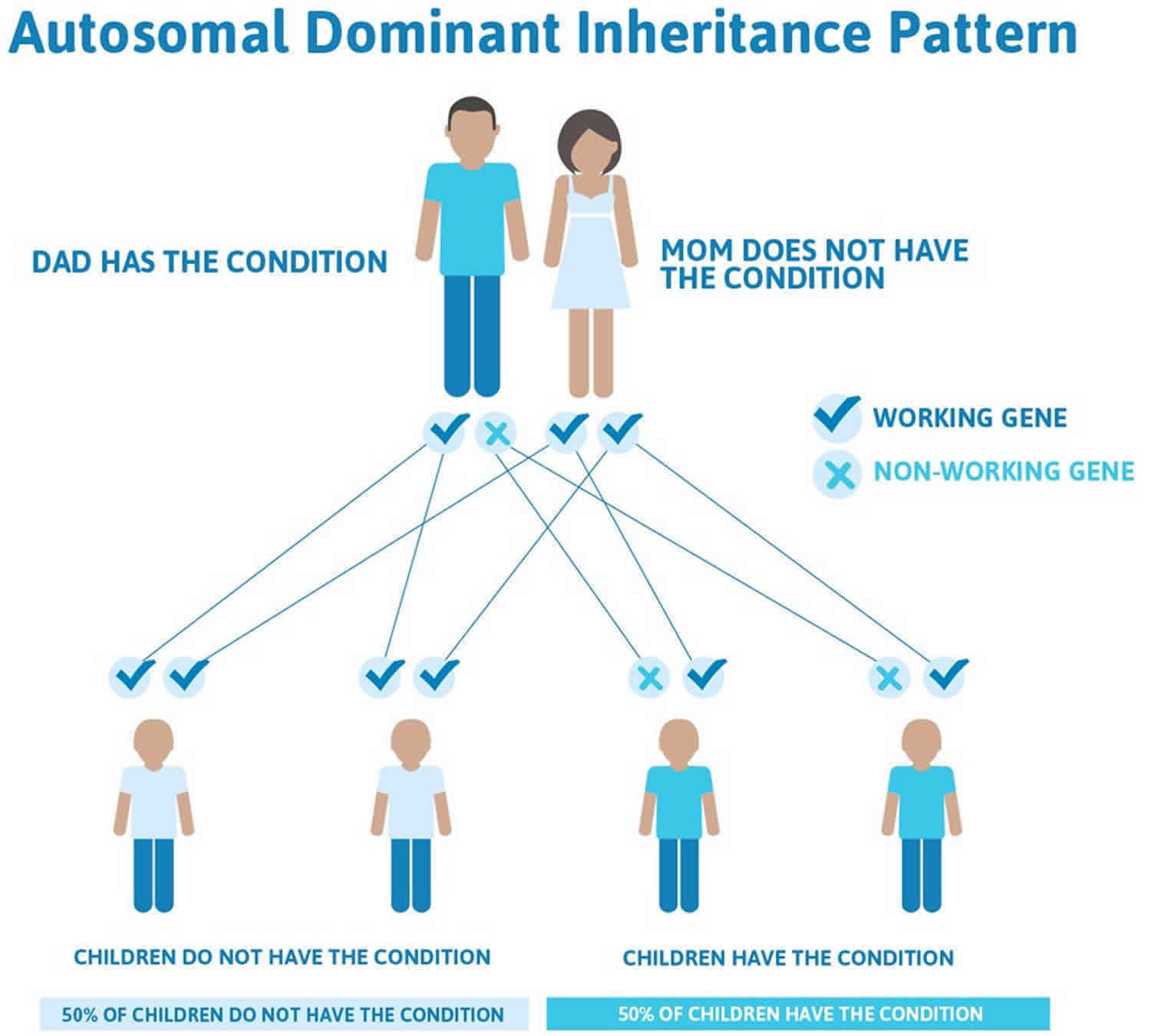
Alexander disease inheritance pattern
Alexander disease is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. The risk of transmitting Alexander disease from an affected parent to an offspring is 50 percent for each pregnancy. The risk is the same for males and females.
Most Alexander disease patients have a new mutation in the GFAP gene, indicating that neither of their parents has the mutation, but the mutation arose at some point during the development of sperm or ova or an embryo. As Alexander disease becomes better diagnosed, familial cases, in which the disease is passed from one generation to the next, are being increasingly recognized. Rarely, an affected person inherits the mutation from one affected parent.
Figure 2. Alexander disease autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Alexander disease symptoms
The symptoms of Alexander disease vary depending on the form of the condition (neonatal, infantile, juvenile, and adult). Even within the different forms there may be huge differences in respect to symptoms and severity 5):
- Neonatal Alexander disease – Leads to severe disability or death within two years. Characteristics include seizures, hydrocephalus, severe motor and intellectual disability.
- Infantile Alexander disease – The most common type of Alexander disease. It has an onset during the first two years of life. Usually there are both mental and physical developmental delays, followed by the loss of developmental milestones, an abnormal increase in head size, and seizures.
- Juvenile Alexander disease – Less common and has an onset between the ages of two and thirteen. These children may have excessive vomiting, difficulty swallowing and speaking, poor coordination, and loss of motor control.
- Adult Alexander disease – Rare and is generally the most mild. Onset can be anywhere from the late teens to very late in life. In some cases the symptoms mimic those of Parkinson disease or multiple sclerosis.
Historically, three forms of Alexander disease have been described based on age of onset, Infantile, Juvenile and Adult; but an analysis of a large number of patients concluded that Alexander disease is better described as having two forms, Type 1, which generally has an onset by age 4, and Type 2, which can have onset at any age, but primarily after age 4 6). Each type accounts for about half of the reported patients. Symptoms associated with the Type 1 form include a failure to grow and gain weight at the expected rate (failure to thrive); delays in the development of certain physical, mental, and behavioral skills that are typically acquired at particular stages (psychomotor impairment); and sudden episodes of uncontrolled electrical activity in the brain (seizures). Additional features typically include progressive enlargement of the head (macrocephaly); abnormally increased muscle stiffness and restriction of movement (spasticity); lack of coordination (ataxia); and vomiting and difficulty swallowing, coughing, breathing or talking (bulbar and pseudobulbar signs). Nearly 90% of infantile patients display developmental problems and seizures, and over 50% the other symptoms mentioned; however, no single symptom or combination of symptoms is always present.
Patients with type 2 Alexander disease rarely show delay or regression of development, macrocephaly or seizures, and mental decline may develop slowly or not at all. Instead, about 50% display bulbar/pseudobulbar signs, about 75% have ataxia and about 33% spasticity. Because these symptoms are not specific, adult Alexander disease is sometimes confused with more common disorders such as multiple sclerosis or the presence of tumors.
The two different forms of Alexander disease are generalizations rather than defined entities. In actuality there is an overlapping continuum of presentations; a one year old could present with symptoms more typical of a 10 years old, and vice-versa. However, in all cases the symptoms almost always worsen with time and eventually lead to death, with the downhill course generally (but not always) being swifter the earlier the onset.
Alexander disease diagnosis
For many years a brain biopsy to determine the presence of Rosenthal fibers was required for the diagnosis of Alexander disease. However, even this procedure can be ambiguous, because Rosenthal fibers are also found in certain other disorders, such as tumors of astrocytes. More recently, MRI criteria have been developed that have a high degree of accuracy for diagnosing typical Type I (early onset) disease. These criteria have been less useful for some of the Type II cases, which have little or no white matter deficits in the brain, although abnormalities in the brainstem, cerebellum, and spinal cord can suggest the diagnosis. Accordingly, when making a diagnosis of Alexander disease, more common diseases that have similar symptoms for which tests are available should first be ruled out. These include adrenoleukodystrophy, Canavan’s disease, glutaricacidurias, Krabbe leukodystrophy, Leigh syndrome, metachromic leukodystrophy, Pelizaeus-Merzbacher and Tay-Sachs disease. A definitive diagnosis of Alexander disease rests on the identification of a GFAP mutation in the patient’s DNA, which can be obtained from a blood sample or a swab of the inside of the cheek. DNA analysis is provided by several commercial and research laboratories. However, since no GFAP mutation has been found in about 5% of known cases, a negative result does not completely rule out the disease. Presently, Alexander patients without a GFAP mutation can be definitively diagnosed only at autopsy by the presence of disseminated, large numbers of Rosenthal fibers.
Alexander disease treatment
There is no cure for Alexander disease, nor is there a standard course of treatment 7). Treatment of Alexander disease is symptomatic and supportive.
Genetic counseling may be of benefit for patients and their families. Fetal diagnosis is an option for a couple who have had a previously affected child.
Current research on Alexander disease is focused on identifying the genetic change in all cases and investigating the mechanism of how the mutations in the GFAP gene lead to the disease. Also being investigated is the exact composition of the Rosenthal fibers and the factors responsible for their formation and growth. Research is also underway to try to find ways to prevent the mutant GFAP from being made or accumulating. Together, these studies may eventually lead to new methods of diagnosis and, in time, to the development of new treatments for Alexander disease.
Alexander disease prognosis
The prognosis for individuals with Alexander disease is generally poor and typically depends of the specific form. People with the neonatal form usually have the worst prognosis. Most children with the infantile form do not survive past the age of 6. The juvenile and adult forms of the disorder have a slower, more lengthy course. The adult form varies greatly and, in some cases, there are no symptoms 8).
Alexander disease life expectancy
Alexander disease life expectancy depends on the specific form.
Infantile Alexander disease leads to symptoms in the first two years of life; while some children die in the first year of life, a larger number live to be 5-10 years old. The usual course of infantile Alexander disease is progressive, leading to eventual severe mental retardation and spastic quadriparesis (spasms that may involve all four limbs). However, in some children the degree of disability develops slowly over several years, and some children retain responsiveness and emotional contact until near the end of their lives. Feeding often becomes a problem, and assisted feeding (as with a nasogastric tube) may become necessary. Their head circumference is often enlarged. Children with hydrocephalus caused by Alexander disease usually have increased intracranial pressure and a more rapid progression of the disease. Generally, the earlier the age of onset of Alexander disease, the more severe and rapid the course.
Juvenile Alexander disease is characterized by difficulty with talking and swallowing and the inability to cough. There can also be weakness and spasticity of the extremities, particularly the legs. Unlike in the infantile form of the disease, mental ability and head size may be normal. Age of onset is usually between the ages of 4 and 10. Survival can extend several years following onset of symptoms, with occasional longer survival into middle age.
Adult-onset Alexander disease is the most rare of the forms, and also is generally the most mild. Onset can be anywhere from the late teens to very late in life. In older patients ataxia (impaired coordination) often occurs and difficulties in speech articulation, swallowing, and sleep disturbances may occur. Symptoms can be similar to those in juvenile disease, although the disease may also be so mild that symptoms are not even noticed until an autopsy reveals the presence of the Rosenthal fibers. Symptoms may resemble multiple sclerosis or a tumor.
References [ + ]




{kind=link}