Contents
Smith Lemli Opitz syndrome
Smith-Lemli-Opitz syndrome is a variable genetic disorder (multiple congenital anomaly or intellectual disability syndrome) that affects many parts of the body caused by a defect in cholesterol synthesis 1). Smith-Lemli-Opitz syndrome is caused by a mutation in the DHCR7 (7-dehydrocholesterol reductase) gene on chromosome 11. This gene codes for a 3 beta-hydroxysterol-delta 7-reductase enzyme (7-dehydrocholesterol-delta 7-reductase [DHCR7]) the final enzyme in the sterol synthetic pathway that converts 7-dehydrocholesterol (7DHC) to cholesterol. People who have Smith-Lemli-Opitz syndrome are unable to make enough cholesterol to support normal growth and development.
Cholesterol is an essential component of the cell membrane and tissues of the brain. A person who can’t make enough cholesterol will therefore experience poor growth, developmental delays, and mental retardation. People with Smith-Lemli-Opitz syndrome may also have a range of physical malformations (such as extra fingers or toes) and problems with internal organs (such as the heart or kidney).
Smith-Lemli-Opitz syndrome is characterized by distinctive facial features, small head size (microcephaly), intellectual disability or learning problems, and behavioral problems. Many affected children have the characteristic features of autism, a developmental condition that affects communication and social interaction. Malformations of the heart, lungs, kidneys, gastrointestinal tract, and genitalia are also common. Infants with Smith-Lemli-Opitz syndrome have weak muscle tone (hypotonia), experience feeding difficulties, and tend to grow more slowly than other infants. Most affected individuals have fused second and third toes (syndactyly), and some have extra fingers or toes (polydactyly).
The signs and symptoms of Smith-Lemli-Opitz syndrome vary widely. Mildly affected individuals may have only minor physical abnormalities with learning and behavioral problems. Severe cases can be life-threatening and involve profound intellectual disability and major physical abnormalities.
Affected individuals usually have low plasma cholesterol levels and invariably have elevated levels of cholesterol precursors, including 7DHC (7-dehydrocholesterol). The most severely affected individuals (those with the condition formerly referred to as Smith-Lemli-Opitz syndrome type II) have multiple congenital malformations and are often miscarried or stillborn or die in the first weeks of life. Dysmorphic facial features, microcephaly, second-toe and third-toe syndactyly, other malformations, and intellectual disability are typical. Mildly affected individuals may have only subtle dysmorphic features and, often, learning and behavioral disabilities.
Smith-Lemli-Opitz syndrome affects an estimated 1 in 20,000 to 60,000 newborns 2). Smith-Lemli-Opitz syndrome is most common in whites of European ancestry, particularly people from Central European countries such as Slovakia and the Czech Republic. Smith-Lemli-Opitz syndrome is very rare among African and Asian populations.
Smith-Lemli-Opitz syndrome is usually suspected clinically, but biochemical studies (and/or genetic studies) are necessary for diagnosis. Currently, no treatment has proven effective long-term for patients with the Smith-Lemli-Opitz syndrome 3). With the right medical care and proper diet a person with Smith-Lemli-Opitz syndrome can experience a normal life expectancy, although independent living is unlikely due to mental retardation. Sadly, children with the most severe cases of Smith-Lemli-Opitz syndrome (produce almost no cholesterol) often die a few months after birth.
Figure 1. Smith-Lemli-Opitz syndrome
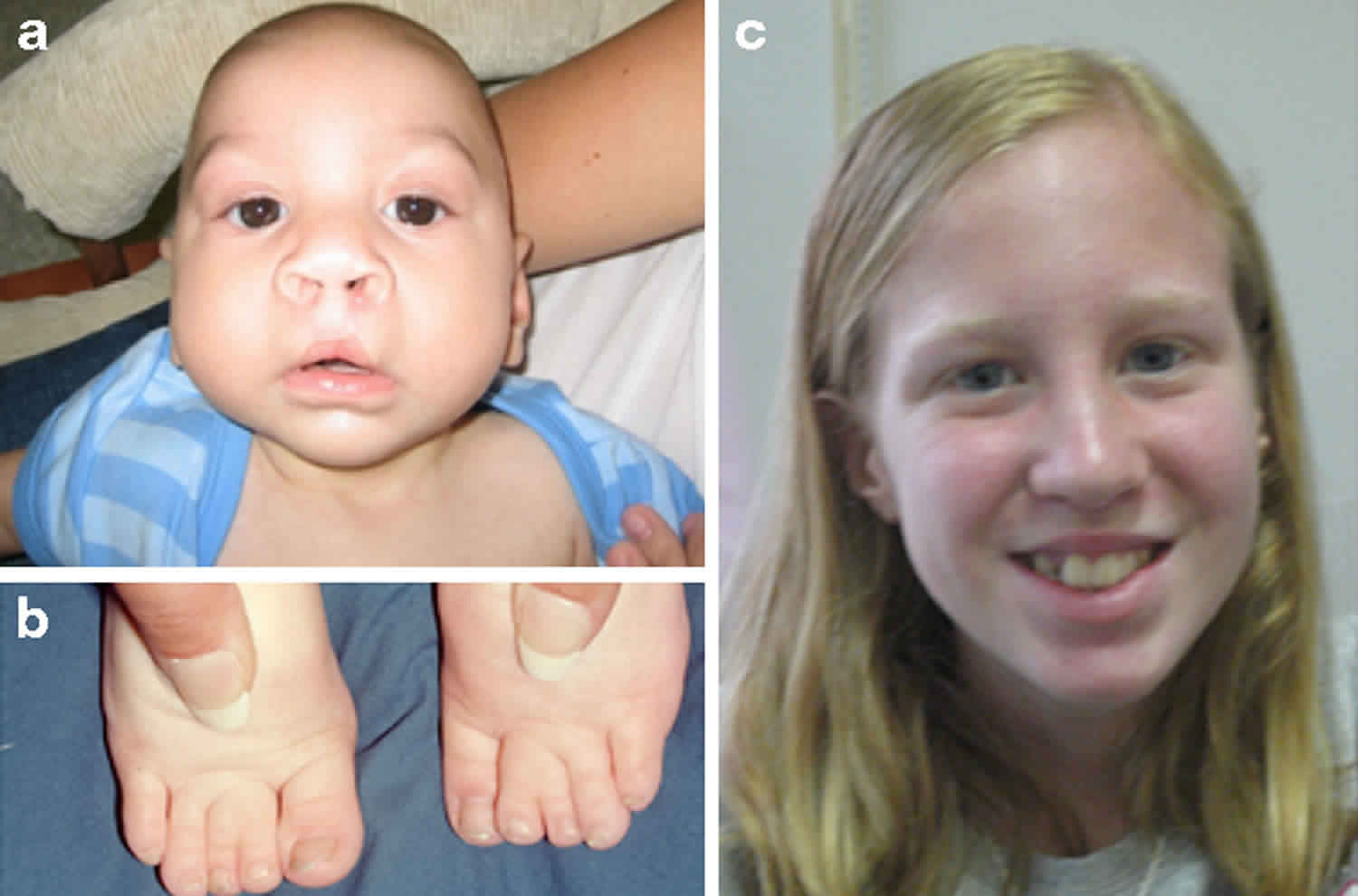
Footnote: Clinical features of Smith–Lemli–Opitz syndrome. (a) Typical facial features of Smith-Lemli-Opitz syndrome: microcephaly, bitemporal narrowing, ptosis, short nasal root, anteverted nares and micrognathia. (b) Toe syndactyly in Smith-Lemli-Opitz syndrome. (c) Smith-Lemli-Opitz syndrome with mild phenotype.
[Source 4) ]Smith-Lemli-Opitz syndrome causes
Mutations in the DHCR7 gene cause Smith-Lemli-Opitz syndrome. The DHCR7 gene provides instructions for making an enzyme called 7-dehydrocholesterol reductase. This enzyme is responsible for the final step in the production of cholesterol. Cholesterol is a waxy, fat-like substance that is produced in the body and obtained from foods that come from animals (particularly egg yolks, meat, poultry, fish, and dairy products). Cholesterol is necessary for normal embryonic development and has important functions both before and after birth. It is a structural component of cell membranes and the protective substance covering nerve cells (myelin). Additionally, cholesterol plays a role in the production of certain hormones and digestive acids.
Mutations in the DHCR7 gene reduce or eliminate the activity of 7-dehydrocholesterol reductase, preventing cells from producing enough cholesterol. A lack of this enzyme also allows potentially toxic byproducts of cholesterol production to build up in the blood, nervous system, and other tissues. The combination of low cholesterol levels and an accumulation of other substances likely disrupts the growth and development of many body systems. It is not known, however, how this disturbance in cholesterol production leads to the specific features of Smith-Lemli-Opitz syndrome.
Smith-Lemli-Opitz syndrome inheritance pattern
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
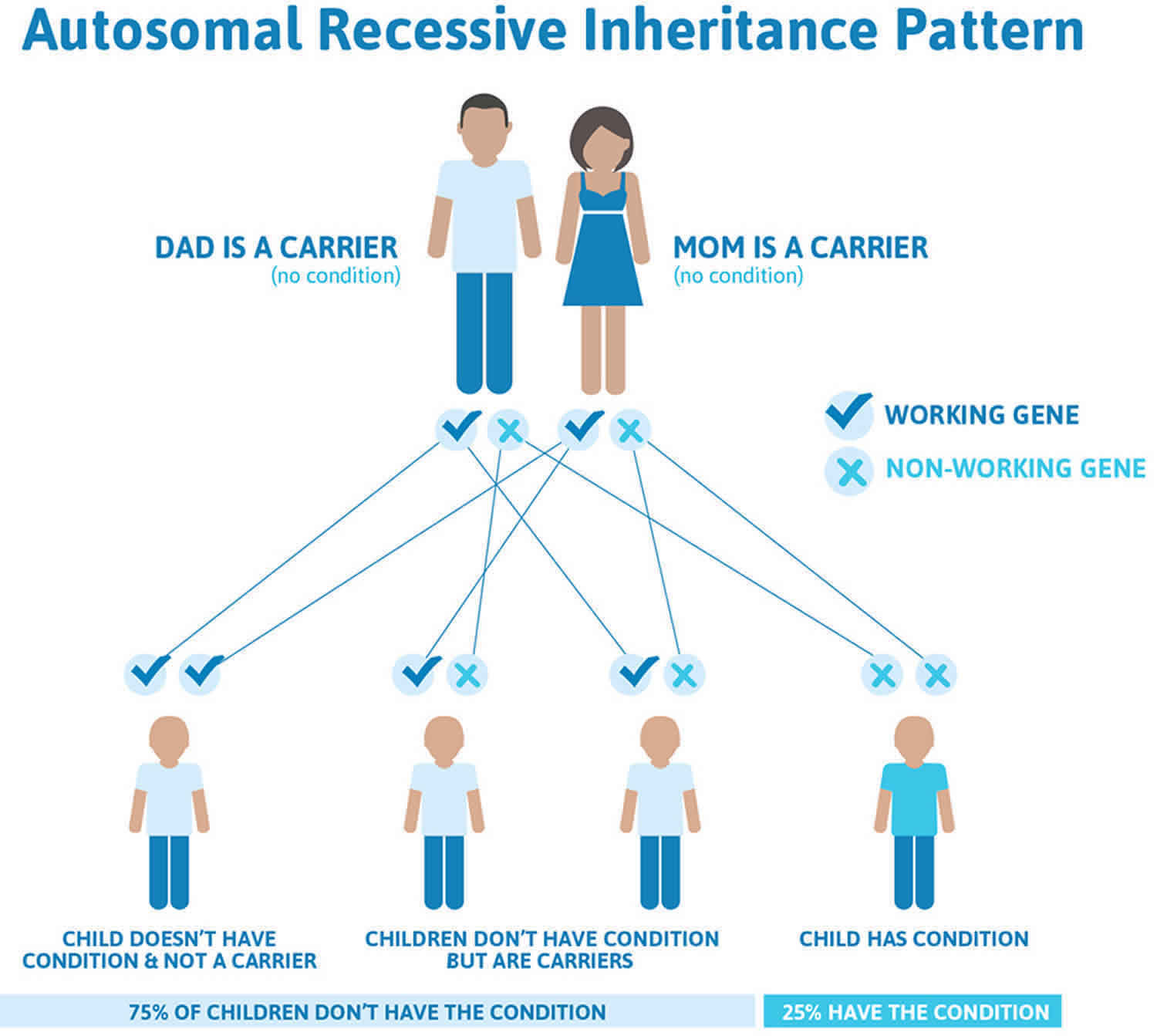
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Smith-Lemli-Opitz syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Smith-Lemli-Opitz syndrome symptoms
Smith-Lemli-Opitz syndrome symptoms vary from person to person, depending upon the amount of cholesterol they can produce. In addition to mental retardation and poor growth, common physical signs of Smith-Lemli-Opitz syndrome are a cleft palate (a split upper lip), malformed genitals (in males), and polydactyly (extra fingers or toes).
Other symptoms that may be present at birth include: microcephaly (small head), webbing between the second and third toes, drooping eyelids, heart defects, hearing or sight loss, and difficulties feeding.
The following signs and symptoms may be noted in Smith-Lemli-Opitz syndrome:
- Lethargy
- Obtundation or coma
- Respiratory failure
- Hearing loss
- Visual loss
- Vomiting
- Feeding difficulties
- Failure to thrive
- Constipation
- Cyanosis
- Congestive heart failure
- Photosensitivity
Neuropsychiatric and neurodevelopmental abnormalities are common and include variable intellectual disability, aberrant behavior, and autism.
Smith-Lemli-Opitz syndrome diagnosis
The diagnosis of Smith-Lemli-Opitz syndrome is based on physical findings and detection of an elevated concentration of 7-dehydrocholesterol (7-DHC) in blood serum or an elevated 7-dehydrocholesterol:cholesterol ratio. Molecular genetic testing for mutations in the DHCR7 gene is available and is mainly used for carrier testing and prenatal diagnosis.
Smith-Lemli-Opitz syndrome treatment
There is no cure for Smith-Lemli-Opitz syndrome. Cholesterol therapy, which comes in several forms and can improve growth and development, is the recommended treatment. The results, however, vary and not every family sees significant change. Other possible treatments such as simvastatins and antioxidants are currently being investigated through clinical trials. Simvastatin has been successful in reducing cholesterol levels in individuals with Smith-Lemli-Opitz syndrome but must be used with caution because of the possible risk of liver damage. Surgery may be necessary to correct some of the physical deformities (cleft palate, heart defects) associated with the disorder.
Medical treatment for Smith-Lemli-Opitz syndrome is based on the specific problems that are present in the affected child. It is important that the child be evaluated for the range of conditions associated with Smith-Lemli-Opitz syndrome including eye, heart, musculoskeletal, genitourinary and gastrointestinal disorders and that a physician familiar with Smith-Lemli-Opitz syndrome oversees the care. Severely affected individuals may require surgery to correct cleft palate, heart defects and genital anomalies. Cholesterol supplementation (one or two egg yolks), sometimes in combination with bile acids, appears to improve growth and reduce photosensitivity in individuals with Smith-Lemli-Opitz syndrome with no harmful side effects.
Genetic counseling is recommended for the parents of an affected child.
Surgical care
- Consider repair of congenital heart defects in cases of Smith-Lemli-Opitz syndrome type I.
- Repair of polydactyly is best performed early.
- Consider cleft palate repair as well as pyloromyotomy in a timely fashion in cases of pyloric stenosis.
- Rectal biopsy for evaluation of ganglion cells may be useful when Hirschsprung disease is suspected and surgical management for Hirschsprung disease may be needed.
- Gastrostomy placement, with or without fundoplication, may be necessary when feeding difficulties or gastrointestinal reflux is present.
Smith-Lemli-Opitz syndrome life expectancy
A person with Smith-Lemli-Opitz syndrome who has appropriate medical care and follows a proper diet has the potential for a normal life expectancy. Independent living is unlikely, however, due to the presence of intellectual disability. Children with the most severe cases of Smith-Lemli-Opitz syndrome (those who produce little or no cholesterol) often die within a few months of birth 5).
References [ + ]




{kind=link}