Contents
What is tricuspid atresia
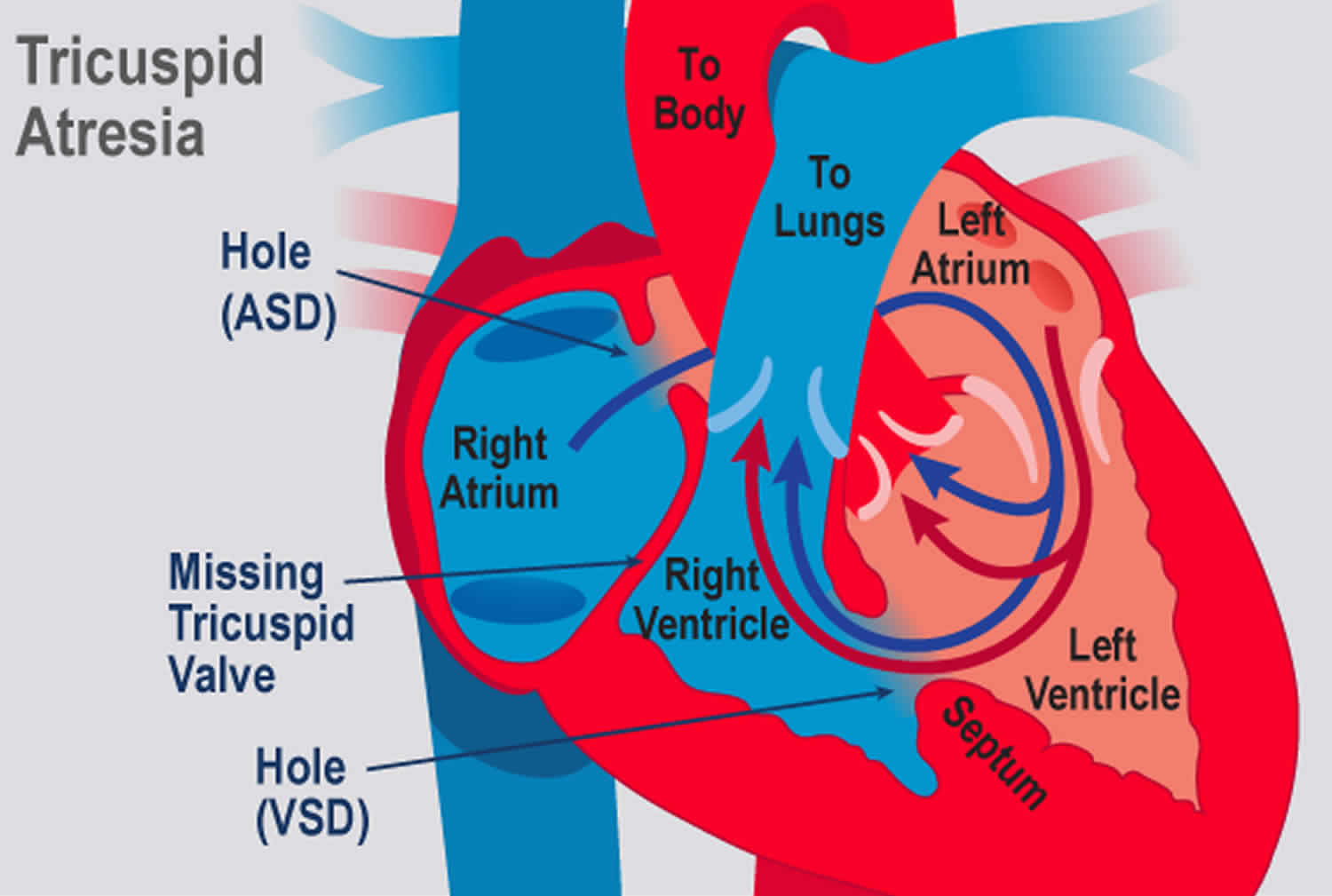
Tricuspid atresia is a congenital (present at birth) heart defect, that occurs when the tricuspid heart valve is missing or abnormally developed. The defect blocks blood flow from the right atrium (upper receiving chamber) to the right ventricle (lower pumping chamber) (see Figure 2). Ultimately blood cannot enter the lungs, where it must go to pick up oxygen. A baby born with tricuspid atresia often has serious symptoms soon after birth because blood flow to the lungs is much less than normal.
The symptoms of tricuspid atresia vary from one baby to another. Some children look remarkably well while some children become blue if there is too little blood flowing to the lungs. Others become breathless early in life if there is too much blood flowing to the lungs. Children who are breathless may not gain weight normally.
Normally, blood flows from the body into the right atrium, then through the tricuspid valve to the right ventricle and on to the lungs (see Figure 1). If the tricuspid valve does not open, the blood cannot flow from the right atrium to the right ventricle. Because of the problem with the tricuspid valve, blood ultimately cannot enter the lungs. This is where it must go to pick up oxygen (becomes oxygenated).
Instead, the blood passes through a hole between the right and left atrium. In the left atrium, it mixes with oxygen-rich blood returning from the lungs. This mix of oxygen-rich and oxygen-poor blood is then pumped out into the body from the left ventricle. This causes the oxygen level in the blood to be lower than normal.
In people with tricuspid atresia, the lungs receive blood either through a hole between the right and left ventricles (described above), or through maintenance of a fetal vessel called the ductus arteriosus. The ductus arteriosus connects the pulmonary artery (artery to the lungs) to the aorta (main artery to the body). It is present when a baby is born, but normally closes by itself shortly after birth.
Contact your health care provider right away if your infant has:
- New changes in breathing patterns
- Problems eating
- Skin that is turning blue
Figure 1. Normal heart valves
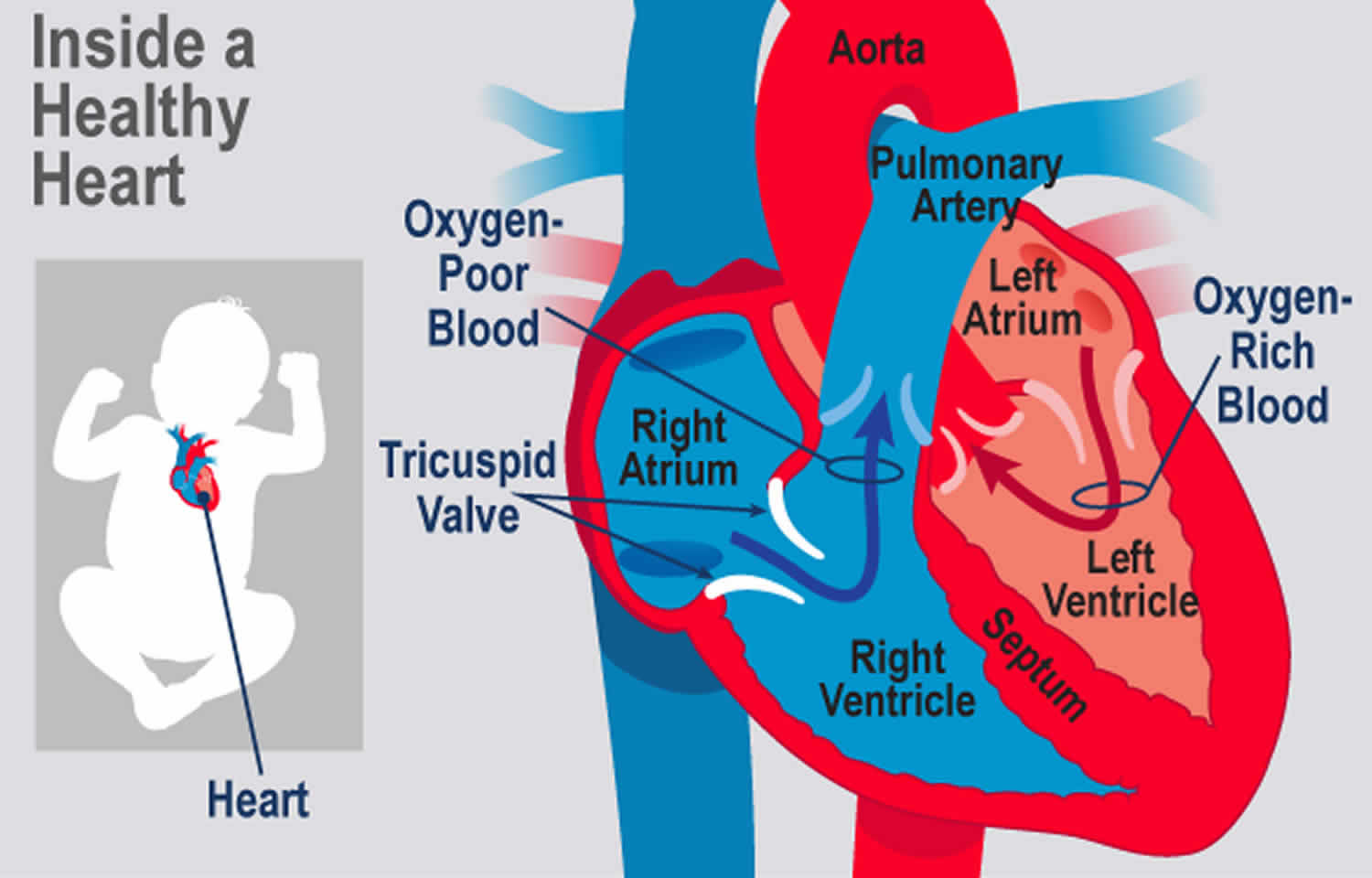
Figure 2. Tricuspid valve atresia
What happens as my child grows up?
Tricuspid atresia is a complex condition, and even with surgical treatment it cannot be corrected. Children with this condition are often limited to some extent in their physical activities, but specific restrictions on exercise are not always necessary. Your child’s cardiologist will tell you if there are any specific forms of exercise or activities they should avoid.
Although surgery can give a better quality of life, it is not possible to correct the heart abnormality and it’s uncertain how long children with this condition will live for. The longest survivors at present are in their later 30s. Heart transplantation may be an option for some patients, although this is rarely considered before adulthood.
Adults with tricuspid atresia
If you’re an adult with tricuspid atresia, you need to be seen regularly throughout your life by a doctor trained in adult congenital heart conditions. Your doctor is likely to recommend regular tests to evaluate your condition at these appointments.
Your doctor might recommend that you take preventive antibiotics before certain dental or medical procedures to prevent infective endocarditis.
Ask your doctor about what activities are best for you, and if there are sports or activities that you should limit or avoid.
Tricuspid atresia and pregnancy
Women with tricuspid atresia who are considering pregnancy should talk to a doctor who specializes in adult congenital heart diseases as well as a maternal-fetal medicine specialist. If you do become pregnant, it’s best to see a doctor who specializes in pregnancies in women with congenital heart disease.
For women who have had a Fontan procedure, pregnancy will be considered high-risk. Some women, such as those with a history of heart failure, will be discouraged from becoming pregnant.
What is the risk of having another child with a congenital heart condition?
If you have one child with a congenital heart condition, there is around a 1 in 40 chance that if you have another child, they will have a heart condition too 1). However, this risk may be higher (or lower) depending on the type of congenital heart condition your child has. Because your risk of having another child with a congenital heart condition is higher than it is for other people, your doctor may offer you a special scan at an early stage in future pregnancies, to look at the baby’s heart.
Ask your doctor for more information on having a scan earlier than usual. Do be aware that if you have more than one child with a congenital heart condition, the specific condition may not always be the same.
Tricuspid atresia types
Tricuspid atresia is a very serious type of congenital heart disease. There are three main abnormalities:
- The tricuspid valve failed to develop, so there is no connection between the right atrium and the right ventricle.
- The right ventricle is very small
- There is a hole in the ventricular septum (the wall between the ventricles). This is called a ‘ventricular septal defect’ (VSD).
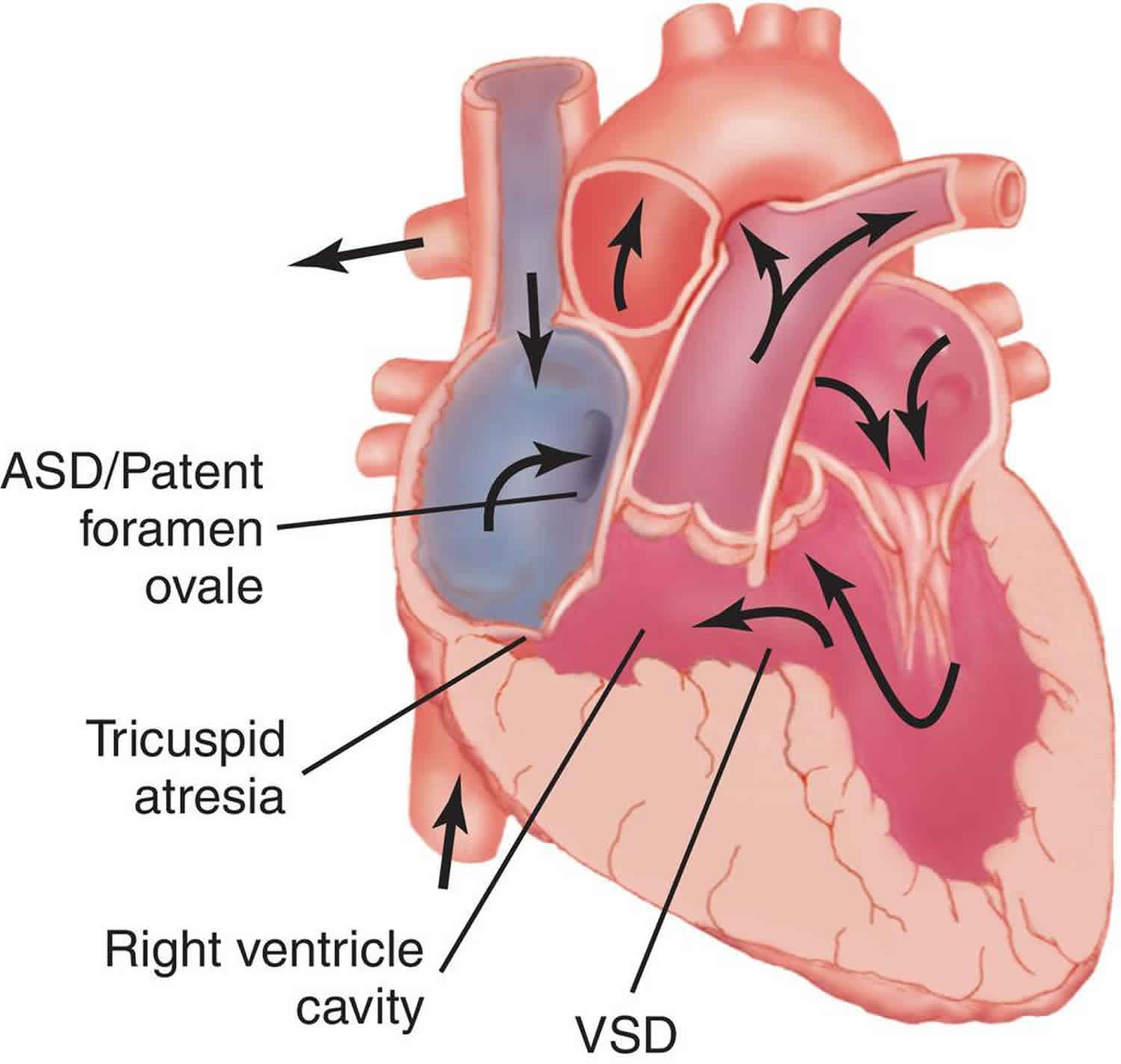
In the normal heart, blood flows from the right atrium through the tricuspid valve to the right ventricle, and from there it goes through the pulmonary artery to the lungs (see Figure 1). In children with tricuspid atresia, the blood cannot flow from the right atrium into the right ventricle (Figure 2). Instead, it flows from the right atrium to the left atrium through a hole in the atrial septum. From the left atrium, the blood flows to the left ventricle, which pumps blood to both the aorta and (through the ventricular septal defect) to the pulmonary artery.
Children with tricuspid atresia may also have other abnormalities of the heart. In some children, the main arteries are ‘transposed’ (transposition of the great arteries). This means that the aorta arises from the right ventricle instead of the left, and the pulmonary artery arises from the left ventricle instead of the right.
In some children there may be a narrowing of the aorta, known as coarctation of the aorta. Others may have pulmonary stenosis (narrowing of the pulmonary valve), or pulmonary atresia (complete blockage of the pulmonary valve). So, some children will have too much blood flowing to the lungs, and some will have too little. Your child’s cardiologist will discuss your child’s individual condition with you.
Tricuspid atresia causes
No one knows why the the tricuspid valve doesn’t grow normally. Tricuspid valve atresia is an uncommon form of congenital heart disease. It affects about 5 in every 100,000 live births. One in 5 people with tricuspid valve atresia will also have other heart problems.
A baby is more likely to have tricuspid atresia if:
- the baby has Down syndrome (trisomy 21)
- either parent has a congenital heart defect
- the mother had a rubella (German measles) infection or other viral infection during pregnancy
- the mother has poorly controlled diabetes or lupus (an autoimmune disease)
- the mother uses certain anti-acne or anti-seizure medicines during pregnancy
But, having one or more risk factors doesn’t mean that a baby will have tricuspid atresia. Tricuspid atresia can happen without any risk factors.
Risk factors for developing tricuspid atresia
In most cases, the cause of a congenital heart defect, such as tricuspid atresia, is unknown. However, several factors might increase the risk of a baby being born with a congenital heart defect, including:
- A mother who had German measles (rubella) or another viral illness during early pregnancy
- A parent who has a congenital heart defect
- Older parental age at conception
- Mother’s obesity
- Drinking alcohol during pregnancy
- Smoking before or during pregnancy
- A mother who has poorly controlled diabetes
- Use of some types of medications during pregnancy, such as the acne drug isotretinoin (Claravis, Amnesteem, others), some anti-seizure medications and some bipolar disorder medications
- The presence of Down syndrome, a genetic condition that results from an extra 21st chromosome
Tricuspid atresia prevention
Congenital heart defects such as tricuspid atresia usually aren’t preventable. If you have a family history of heart defects or a child with a congenital heart defect, a genetic counselor and a cardiologist experienced in congenital heart defects can help you look at risks associated with future pregnancies.
Some steps you can take that might reduce your baby’s risk of heart and other birth defects in pregnancy include:
- Get adequate folic acid. Take 400 micrograms of folic acid daily. This amount, which is often in prenatal vitamins, has been shown to reduce brain and spinal cord defects, and folic acid may help prevent heart defects, too.
- Talk with your doctor about medication use. Whether you’re taking prescription or over-the-counter drugs, an herbal product or a dietary supplement, check with your doctor before using them during pregnancy.
- Avoid smoking or drinking alcohol during pregnancy. Either can increase the risk of congenital heart defects.
- Avoid chemical exposure, whenever possible. While you’re pregnant, it’s best to stay away from chemicals, including cleaning products and paint, as much as you can.
Tricuspid atresia symptoms
Tricuspid atresia symptoms include:
- Bluish color to the skin (cyanosis) due to low oxygen level in the blood
- Fast breathing
- Fatigue
- Have problems feeding
- Get tired quickly when feeding
- Poor growth
- Shortness of breath
- Be less active than most babies
In tricuspid atresia, the right side of the heart can’t pump enough blood to the lungs because the tricuspid valve is missing. A sheet of tissue blocks the flow of blood from the right atrium to the right ventricle. As a result, the right ventricle is usually small and underdeveloped (hypoplastic).
Blood instead flows from the right atrium to the left atrium through a hole in the wall between them (septum). This hole is either a heart defect (atrial septal defect) or an enlarged natural opening that’s supposed to close soon after birth (patent foramen ovale or patent ductus arteriosus). A baby with tricuspid atresia might need medication to keep the natural opening from closing after birth or surgery to create an opening.
Many babies born with tricuspid atresia have a hole between the ventricles (ventricular septal defect). In these cases, some blood can flow through the hole between the left ventricle and the right ventricle, and then blood is pumped to the lungs through the pulmonary artery.
However, the valve between the right ventricle and the pulmonary artery (pulmonary valve) might be narrowed, which can reduce blood flow to the lungs. If the pulmonary valve isn’t narrowed and if the ventricular septal defect is large, too much blood can flow to the lungs, which can lead to heart failure.
Some babies may have other heart defects as well.
Tricuspid atresia possible complications
A life-threatening complication of tricuspid atresia is a lack of oxygen to your baby’s tissues (hypoxemia).
Complications later in life
Although treatment greatly improves the outcome for babies with tricuspid atresia, complications can develop later in life, including:
- Formation of blood clots that can lead to a clot blocking an artery in the lungs (pulmonary embolism) or cause a stroke
- Irregular, fast heart rhythms (arrhythmias)
- Chronic diarrhea (from a disease called protein-losing enteropathy)
- Easy tiring when participating in activity or exercise
- Heart failure
- Fluid in the abdomen (ascites) and in the lungs (pleural effusion)
- Kidney or liver disease
- Blockage of the artificial shunt
- Strokes and other nervous system complications
- Sudden death
Tricuspid atresia diagnosis
Tricuspid atresia may be discovered during routine prenatal ultrasound imaging or when the baby is examined after birth. Bluish skin is present at birth. A heart murmur is often present at birth and may increase in loudness over several months.
A fetal echocardiogram (a more detailed ultrasound study of the unborn baby’s heart) can give more information and help the delivery team plan treatment.
A screening pulse oximeter test usually is done on all newborns right after birth using a light on a fingertip or toe. If tricuspid atresia isn’t found before birth, this test will show that the baby’s blood is not carrying as much oxygen as expected. The delivery team will then do other tests to find the problem and help plan treatment.
Tests may include the following:
- Pulse oximeter monitoring. This measures the oxygen in your or your baby’s blood using a sensor placed over the end of your or your baby’s finger.
- ECG also called EKG, a recording of the heart’s electrical activity
- Echocardiogram (ultrasound images and videos of the heart). This test uses sound waves that bounce off your baby’s heart to produce moving images the doctor can view on a video screen. In a baby with tricuspid atresia, the echocardiogram reveals the absence of a tricuspid valve, irregular blood flow and other heart defects.
- Chest x-ray. This might show whether the heart and its chambers are enlarged. It can also show whether there is too much or too little blood flow to the lungs.
- Cardiac catheterization. A thin, flexible tube (catheter) is inserted into a blood vessel at your child’s groin and guided into the heart. Rarely used to diagnose tricuspid atresia, this test might be used to examine the heart before surgery to treat tricuspid atresia.
- MRI of the heart
- CT scan of the heart
Tricuspid atresia treatment
Once tricuspid atresia diagnosis is made, the baby will often be admitted to the neonatal intensive care unit (NICU). A medicine called prostaglandin E1 may be used to keep the ductus arteriosis open so that blood can circulate to the lungs.
Generally, patients with tricuspid atresia require surgery. If the heart is unable to pump enough blood out to the lungs and rest of the body, the first surgery most often takes place within the first few days of life. In this procedure, an artificial shunt is inserted to keep blood flowing to the lungs. In some cases, this first surgery is not needed.
Afterward, the baby goes home in most cases. The child will need to take one or more daily medicines and be closely followed by a pediatric cardiologist. This doctor will decide when the second stage of surgery should be done.
The next stage of surgery is called the Glenn shunt or hemi-Fontan procedure. This procedure connects half of the veins carrying oxygen-poor blood from the upper half of the body directly to the pulmonary artery. The surgery is most often done when the child is between 4 to 6 months old.
During stage 1 and 2, the child may still look blue (cyanotic).
Stage 3, the final step, is called the Fontan procedure. The rest of the veins carrying oxygen-poor blood from the body are connected directly to the pulmonary artery leading to the lungs. The left ventricle now only has to pump to the body, not the lungs. This surgery is usually performed when the child is 18 months to 3 years old. After this final step, the baby’s skin is no longer blue.
Tricuspid atresia surgery
It’s not possible to correct tricuspid valve atresia with surgery, but there are operations that can help children to have a better quality of life. The type and timing of surgery recommended for a baby with tricuspid atresia will depend on which additional abnormalities they may have, and how severe they are.
These surgical steps (called the single ventricle pathway) can improve blood flow to the lungs in a baby with tricuspid atresia:
- At age 2 weeks or less (Shunt operation): Babies with too little blood flowing to the lungs need surgery to correct it – called a shunt operation. A Blalock-Taussig shunt redirects some of the left ventricle’s output from the body to the lungs. –
- OR – If the blood flow to the lungs is too high, as can happen with a large ventricular septal defect, a band around the pulmonary artery lessens the flow to prevent damage, this surgery is called pulmonary artery banding. The band reduces the high blood flow to the lungs, reducing breathlessness and lowering the blood pressure in the pulmonary artery, to try to prevent lung damage. The surgery usually leaves a scar at the side of the chest rather than in the middle.
- Babies who also have coarctation of the aorta (narrowing of the aorta) usually need surgery to repair the narrowing of the aorta within the first few weeks of life. If your child needs surgery for coarctation of the aorta, the surgeon will place a clamp on the aorta to stop the blood flow and make it easier to operate. He or she will then cut out the narrowed part of the aorta and sew the ends back together. Or, the surgeon may use a patch made of a special material to enlarge the narrowing. After the operation, your child will have a scar either on the left side of the chest or under their arm, or on the middle of the chest.
- At age 4‒6 months: The Glenn Shunt (cavopulmonary shunt) procedure allows blood returning from the upper part of the body to flow directly to the lungs. The Blalock-Taussig shunt is removed at the same time. The Glenn Shunt involves connecting the superior vena cava directly to the pulmonary arteries (the arteries that takes blood to the lungs). The Glenn Shunt (cavopulmonary shunt) procedure procedure is used to increase the blood flow to the lungs, and also to reduce the workload of the heart. A Glenn Shunt (cavopulmonary shunt) does not correct the underlying heart abnormality. Further surgery after this usually involves redirecting the blood flow from the inferior vena cava to the pulmonary artery. This is called a total cavopulmonary connection.
- Most children survive the Glenn Shunt (cavopulmonary shunt) surgery, but they may become more blue and short of breath on exertion as they grow. The risk of death and other complications – such as narrowing where the superior vena cava has been joined to the pulmonary artery, brain damage, stroke or internal bleeding – varies based on the exact type of heart condition your child has. Other possible complications include pleural effusion (fluid around the lungs) and kidney damage. Your pediatric cardiologist or cardiac surgeon will discuss your child’s individual risk with you before surgery.
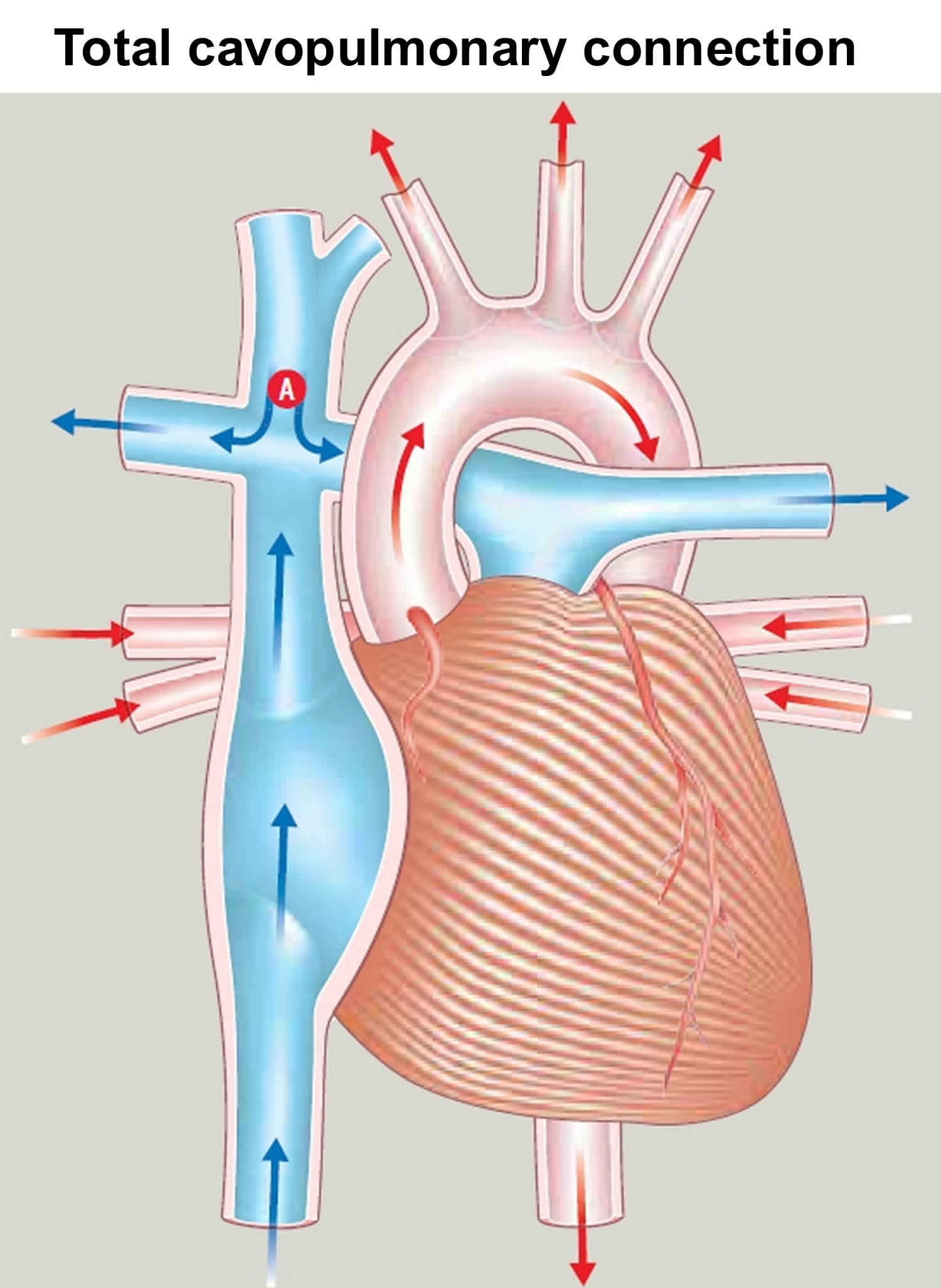
- At age 1.5‒3 years: The Fontan procedure channels blood from the lower half of the body to the lungs so the heart pumps oxygen-rich blood to the body only. Blood returning from the body flows to the lungs before passing through the heart. The purpose of Fontan procedure is to improve the amount of oxygen in the blood and in most cases to improve exercise capacity. This is achieved by connecting both the inferior and superior vena cava to the pulmonary artery. Many modifications to the original Fontan operation technique have been developed, including: a modified Fontan, a fenestrated Fontan, and total cavopulmonary connection. Any type of Fontan operation is a major operation, and your child’s cardiac surgeon will explain exactly which operation your child needs. Your child will be given a general anaesthetic. The heart will be stopped and the heart’s function will be taken over by a heart-lung machine. The surgeon will redirect the flow of blood from the inferior vena cava to the pulmonary artery. In most cases, the superior vena cava has already been connected (see Figure 6). The illustration shows the total cavopulmonary connection type of Fontan operation. After surgery, your child will have a scar in the middle of the chest, along the breastbone. A Fontan-type operation will not make your child’s heart normal, but – if the operation is successful – it should allow an adequate blood supply to the lungs to allow your child to grow.
- Most children survive the Fontan procedure. The risk of death and major complications – such as brain damage – varies depending on the exact type of heart condition your child has. Other possible complications include pleural effusion (fluid around the lungs), pericardial effusion (fluid around the heart), and kidney damage. Some children can develop heart rhythm disturbances which need to be treated with medicines, or less commonly with a pacemaker. The length of time your child will need to stay in hospital will vary, depending on how well he or she recovers after surgery. There is an increased risk of developing a blood clot after the surgery, so most children will need to take either warfarin or aspirin to help prevent this.
Doctors decide which steps to take based on what they learn from all the tests.
Figure 3. Shunt operation
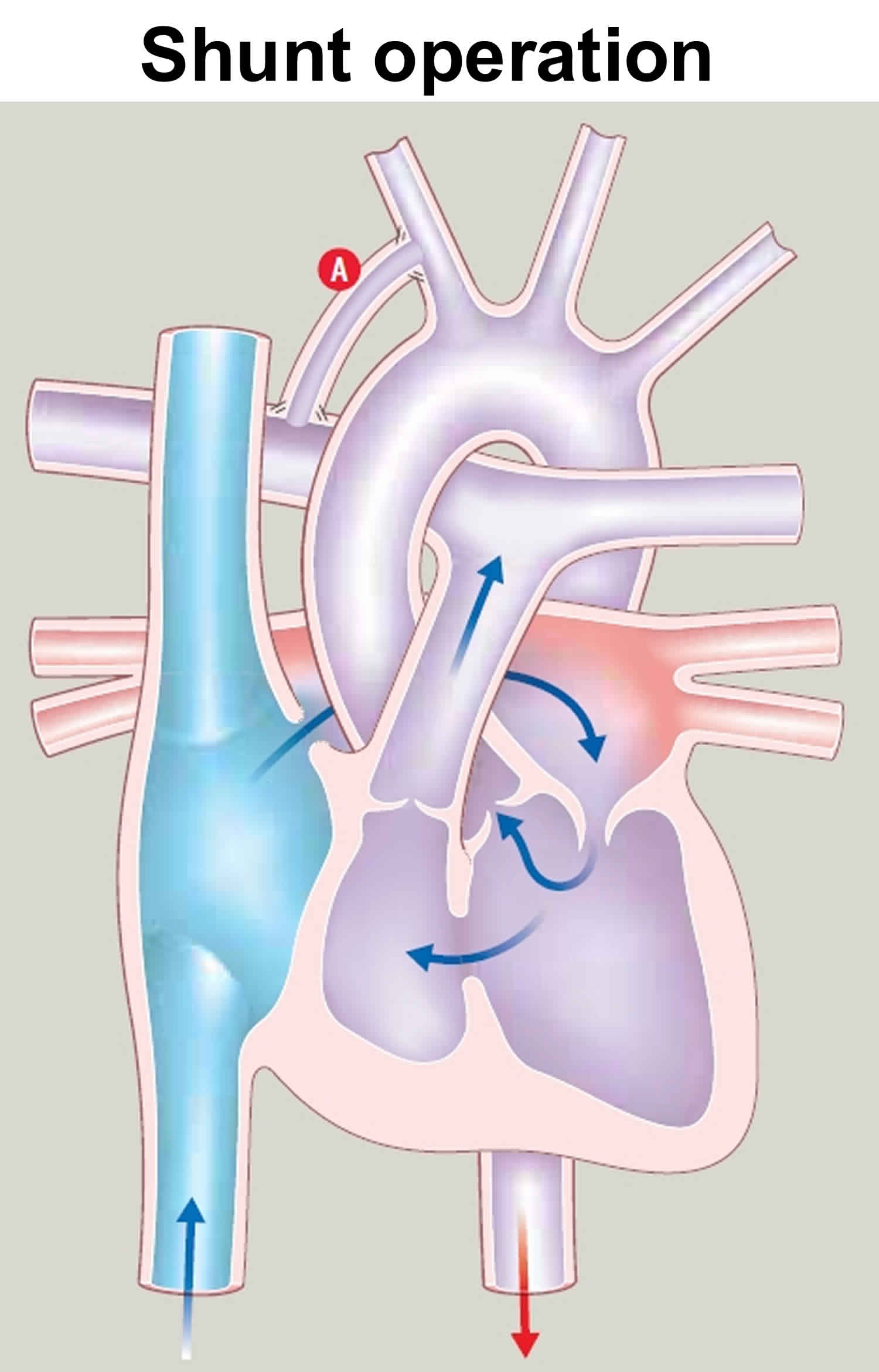
Figure 4. Pulmonary artery banding
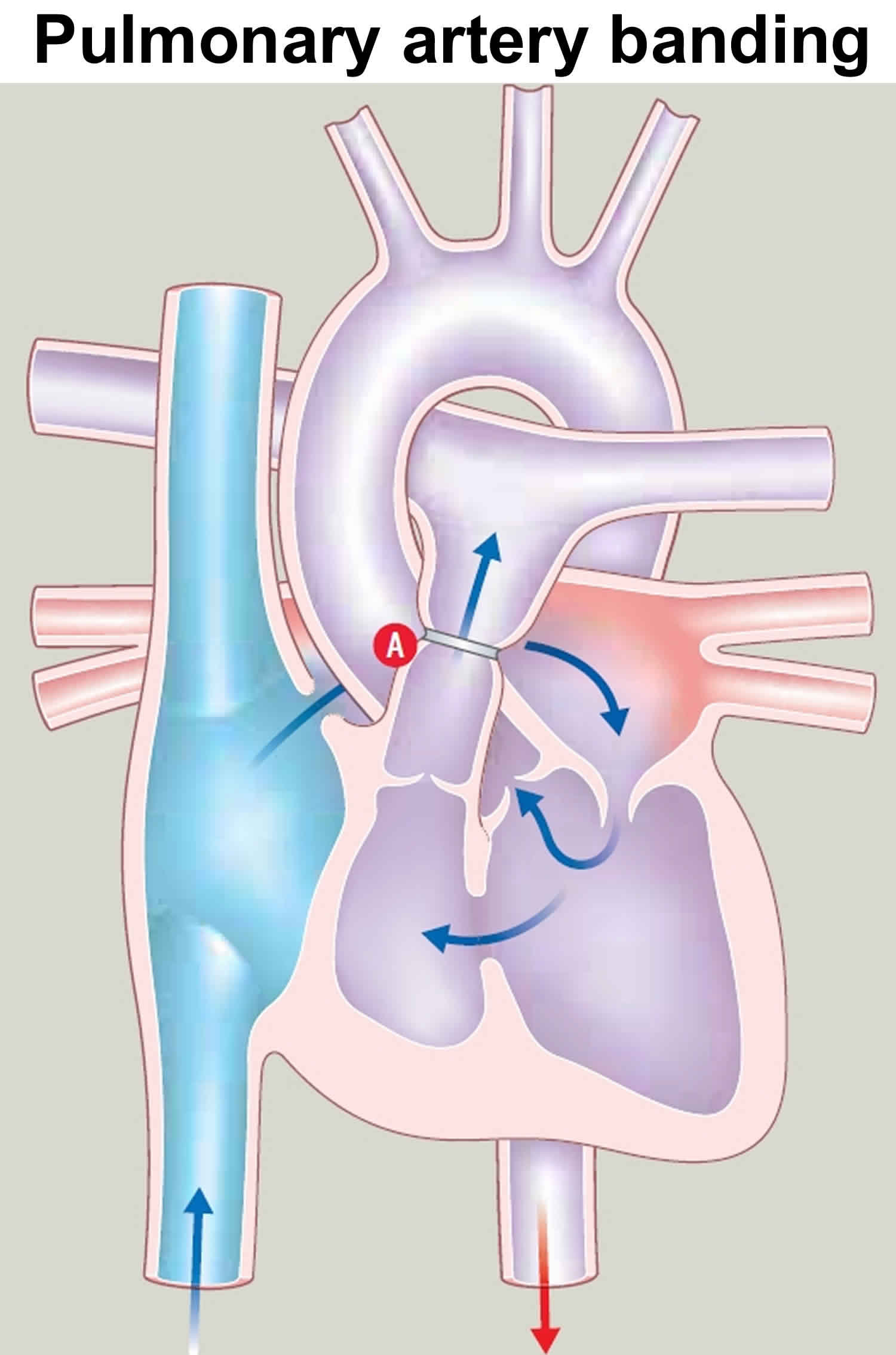
Figure 5. Cavopulmonary shunt (Glenn Shunt) (superior vena cava connected A to pulmonary artery)
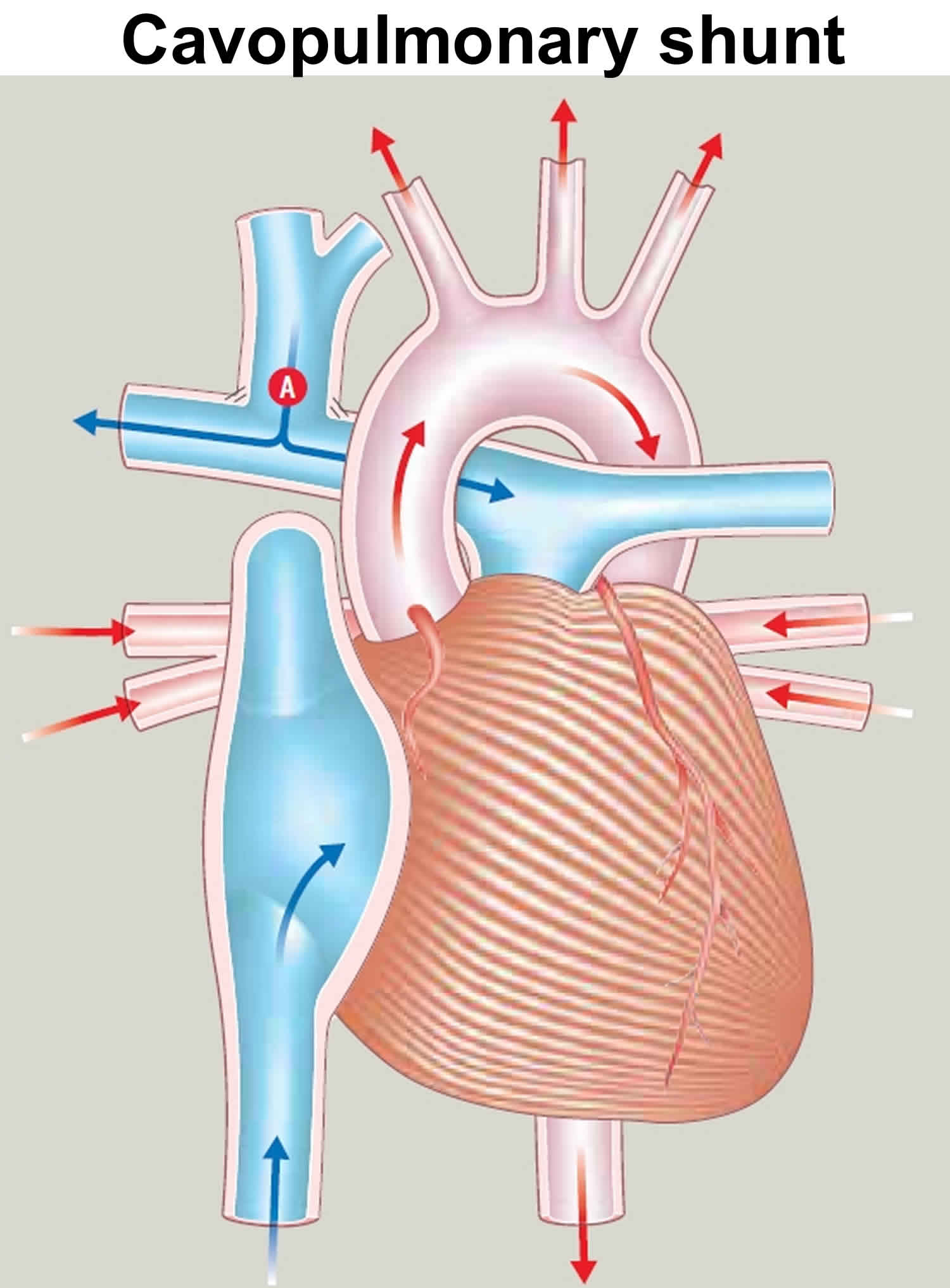
Figure 6. Total cavopulmonary connection (blood flow from both inferior and superior vena cava has been redirected to the right pulmonary artery)
Cardiac Catheterization
Cardiac catheterization can make or enlarge openings in the wall between the two atria and between the two ventricles. It also can be used to place a stent (mesh tube) in the ductus to keep it open.
Lifestyle and home remedies
Here are some tips for caring for your child with tricuspid atresia:
- Strive for good nutrition. Your baby might not be getting enough calories because of tiring during feeding and an increased need for calories. It’s often helpful to give your baby frequent, small feedings. Breast milk is an excellent source of nutrition, but formula works well, too. Your doctor might prescribe a special high-calorie formula.
- Preventive antibiotics. Your or your child’s cardiologist will likely recommend preventive antibiotics be taken before certain dental and other procedures to prevent bacteria from infecting the inner lining of the heart (infective endocarditis).
- Practicing good oral hygiene — brushing and flossing teeth, getting regular dental checkups — also helps prevent infection.
- Stay active. Encourage as much normal play and activity as you or your child can tolerate or as your doctor recommends, with ample opportunity for rest. Staying active helps your or your child’s heart stay fit.
- Keep up with routine medical and well-child care. Standard immunizations are encouraged for children with congenital heart defects, as well as vaccines against the flu, pneumonia and respiratory syncytial virus (RSV) infections. Your child should take all medications as prescribed.
- Keep follow-up appointments with your or your child’s doctor. Your child will need at least annual appointments with a doctor trained in congenital heart conditions. Your child’s doctor is likely to recommend several tests to evaluate your or your child’s heart condition.
Tricuspid atresia prognosis
In most cases, surgery will improve the tricuspid valve atresia. Treatments for tricuspid atresia improve the baby’s condition, but can’t make the heart work like one without a defect. A child born with tricuspid atresia will regularly see a cardiologist who specializes in congenital heart disease. (a doctor who treats heart problems) throughout childhood and as an adult.
Your or your child’s cardiologist will tell you whether you or your child needs to take preventive antibiotics before dental and other procedures. In some cases, your child’s cardiologist might recommend limiting vigorous physical activity.
The short- and intermediate-term outlook for children who have a Fontan procedure is generally promising. A variety of complications can occur over time and require additional monitoring and procedures.
Failure of the circulation system created by the Fontan procedure might make a heart transplant necessary.
Tricuspid atresia life expectancy
Although surgery can give a better quality of life, it is not possible to correct the heart abnormality and it’s uncertain how long children with this condition will live for. The longest survivors at present are in their later 30s. Heart transplantation may be an option for some patients, although this is rarely considered before adulthood.
References [ + ]




{kind=link}