Contents
- What is spherocytosis
- Hereditary spherocytosis
- How might hereditary spherocytosis affect pregnancy?
- How might splenectomy affect pregnancy?
- What are the long term effects of removal of spleen and gallbladder in children with hereditary spherocytosis?
- What are the current recommendations regarding post-splenectomy antibiotic prophylaxis in children?
- Does hereditary spherocytosis increase the risk of stroke or heart attack?
- Hereditary spherocytosis causes
- Hereditary spherocytosis symptoms
- Hereditary spherocytosis complications
- Hereditary spherocytosis prognosis
- Hereditary spherocytosis diagnosis
- Hereditary spherocytosis treatment
- Spherocytosis causes
- Hereditary spherocytosis
What is spherocytosis
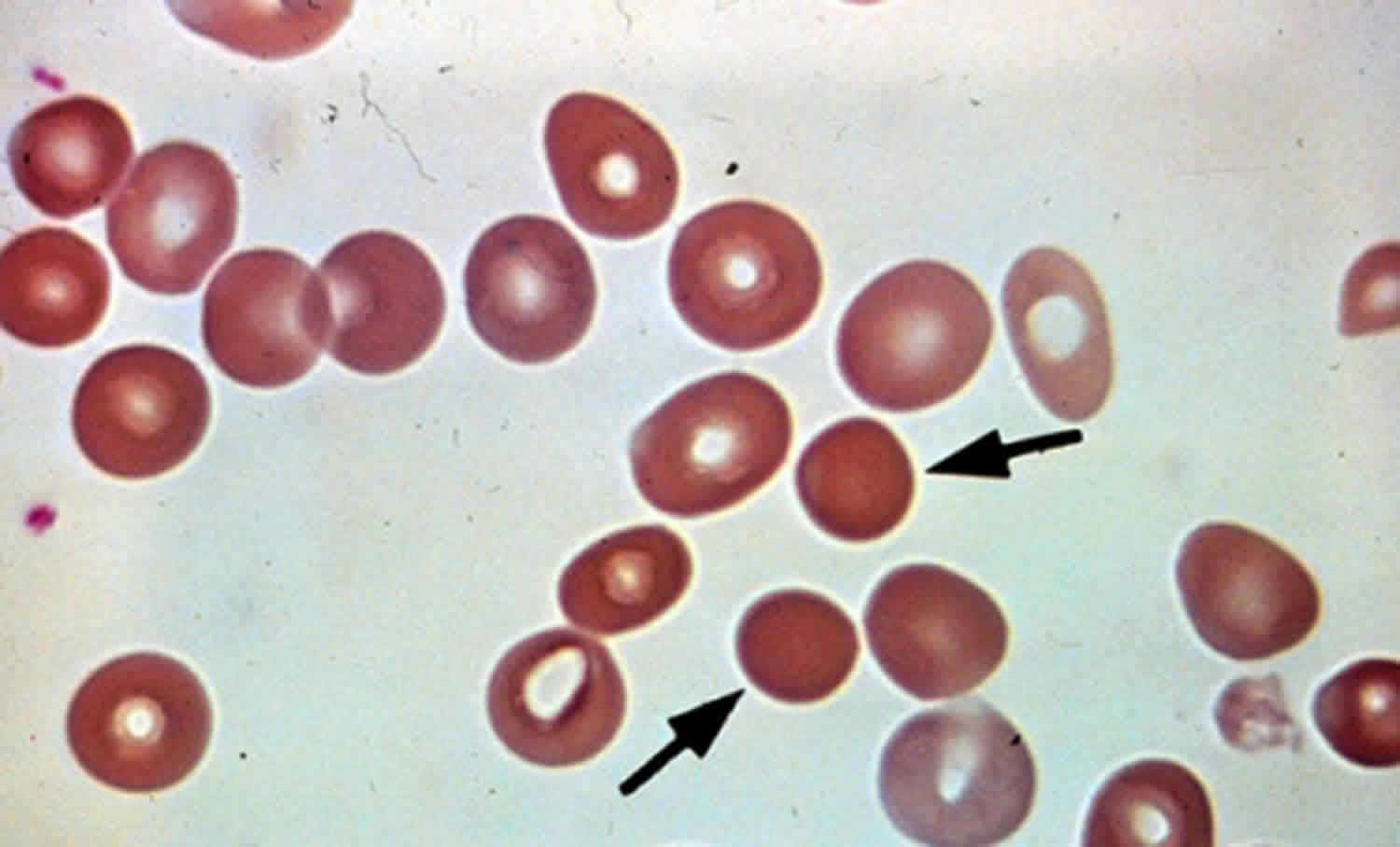
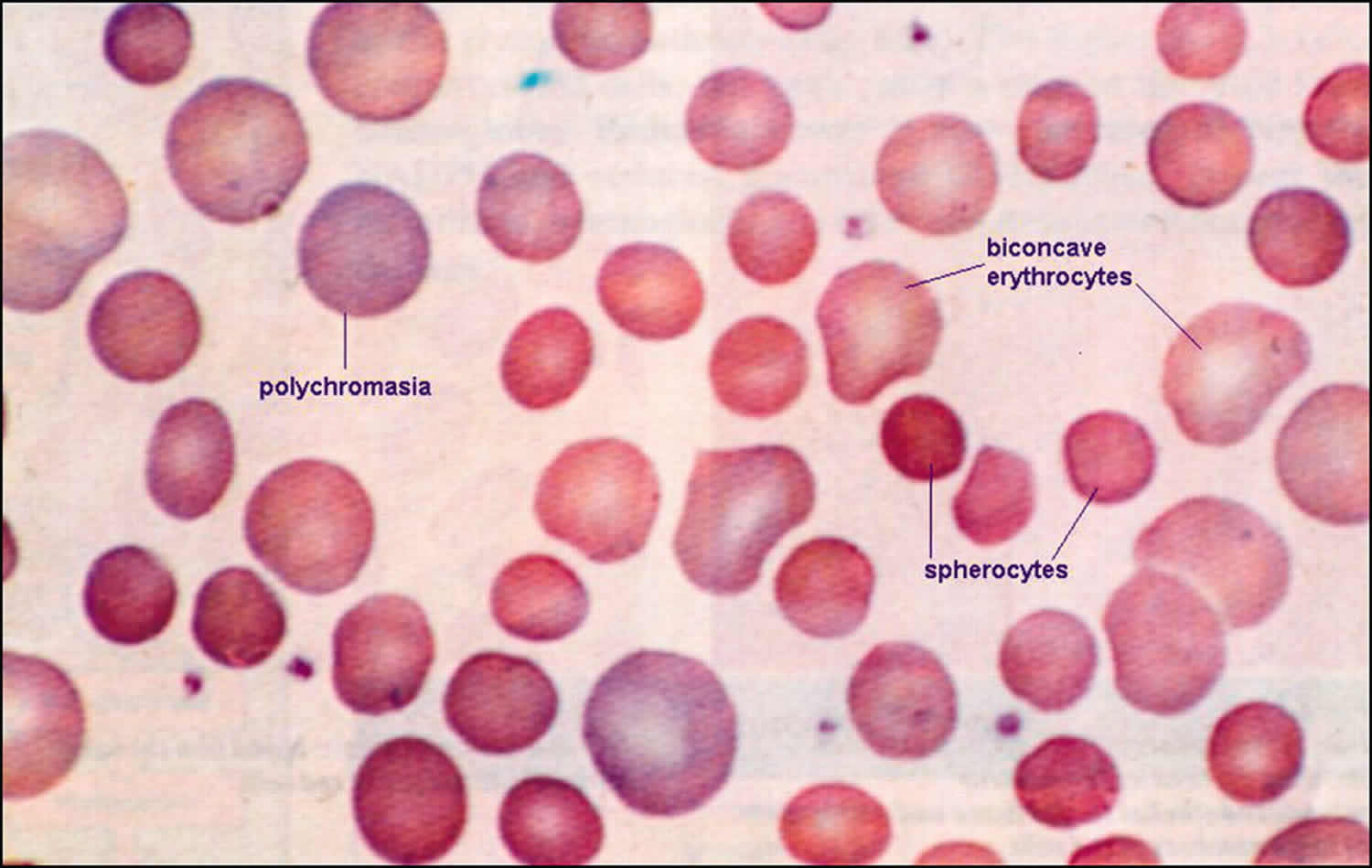
Spherocytosis is the presence in the blood of spherocytes, i.e erythrocytes (red blood cells) that are sphere-shaped rather than bi-concave disk shaped as normal. Spherocytes are found in all hemolytic anemias to some degree. Hereditary spherocytosis and autoimmune hemolytic anemia are characterized by having only spherocytes 1). Spherocytosis most often refers to hereditary spherocytosis. This is caused by a molecular defect in one or more of the proteins of the red blood cell cytoskeleton, including spectrin, ankyrin, Band 3, or Protein 4.2 2). Because the cell skeleton has a defect, the blood cell contracts to a sphere, which is its most surface tension efficient and least flexible configuration. Though the spherocytes have a smaller surface area through which oxygen and carbon dioxide can be exchanged, they in themselves perform adequately to maintain healthy oxygen supplies. However, they have a high osmotic fragility—when placed into water, they are more likely to burst than normal red blood cells. These cells are more prone to physical degradation. Families with a history of spherocytosis should have their children screened for hereditary spherocytosis.
Spherocytosis can be diagnosed in Peripheral blood film by seeing spherical red blood cells rather than biconcave. Because Red blood cells are more prone to lysis in water (because the lack some proteins in their cytoskeleton) Osmotic Fragility test can also be helpful in the diagnosis.
Hereditary spherocytosis
Hereditary spherocytosis is an inherited a disorder of the red cell membrane (cytoskeleton protein deficiency) which results in red blood cells that are fragile causing premature breakdown of red blood cells and anemia (hemolytic anemia) 3). The red blood cells have a normal shape at first – flat discs, like a doughnut without the hole. Over time, small bits of their membranes come off when the cells pass through the spleen. This makes the cells become rounder, like spheres. These rounder red blood cells (spherocytes) are easily destroyed. They have a shorter life than normal red blood cells – as short as 10 to 30 days instead of 100 to 120 days for normal red blood cells.
Red blood cells contain a protein called hemoglobin that carries oxygen around your body, bringing it to cells that need it. Because so many red blood cells are destroyed in spherocytosis, you will have low level of red blood cells. This is called hemolytic anemia. If the anemia is severe, your tissues will get less oxygen than normal.
People with hereditary spherocytosis typically experience a shortage of red blood cells (anemia), yellowing of the eyes and skin (jaundice), and an enlarged spleen (splenomegaly) 4). Most newborns with hereditary spherocytosis have severe anemia, although it improves after the first year of life. Splenomegaly (enlarged spleen) can occur anytime from early childhood to adulthood. About half of affected individuals develop hard deposits in the gallbladder called gallstones, which typically occur from late childhood to mid-adulthood.
The morphologic hallmark of hereditary spherocytosis is the microspherocyte, which is caused by loss of red blood cells membrane surface area and has abnormal osmotic fragility in vitro.
Hereditary spherocytosis occurs in 1 in 2,000 individuals of Northern European ancestry. Hereditary spherocytosis is the most common cause of inherited anemia in that population. The prevalence of hereditary spherocytosis in people of other ethnic backgrounds is unknown, but it is much less common.
There are four forms of hereditary spherocytosis, which are distinguished by the severity of signs and symptoms. They are known as the mild form, the moderate form, the moderate/severe form, and the severe form. It is estimated that 20 to 30 percent of people with hereditary spherocytosis have the mild form, 60 to 70 percent have the moderate form, 10 percent have the moderate/severe form, and 3 to 5 percent have the severe form.
People with the mild form may have very mild anemia or sometimes have no symptoms. People with the moderate form typically have anemia, jaundice, and splenomegaly. Many also develop gallstones. The signs and symptoms of moderate hereditary spherocytosis usually appear in childhood. Individuals with the moderate/severe form have all the features of the moderate form but also have severe anemia. Those with the severe form have life-threatening anemia that requires frequent blood transfusions to replenish their red blood cell supply. They also have severe splenomegaly, jaundice, and a high risk for developing gallstones. Some individuals with the severe form have short stature, delayed sexual development, and skeletal abnormalities.
Surgery to remove the spleen (splenectomy) cures the anemia but does not correct the abnormal cell shape. Splenectomy is the standard treatment for patients with clinically severe hereditary spherocytosis, but can be deferred safely in patients with mild uncomplicated hereditary spherocytosis (hemoglobin level >11 g/dL). After the spleen is removed, the life span of the red blood cell returns to normal. Splenectomy usually results in full control of hereditary spherocytosis, except in the unusual autosomal recessive variant of the disorder 5).
Children should wait until age 5 to have splenectomy because of the infection risk. In mild hereditary spherocytosis cases discovered in adults, it may not be necessary to remove the spleen.
Children and adults should be given a pneumococcal vaccine before spleen removal surgery. They also should receive folic acid supplements. Additional vaccines may be needed based on the person’s history.
Figure 1. Hereditary spherocytosis
How might hereditary spherocytosis affect pregnancy?
There is limited information about the effect of hereditary spherocytosis on pregnancy. Hemolytic crisis and persistent anemia have been reported during pregnancy, especially in women who have not undergone splenectomy 7).
One article reported on 8 patients with hereditary spherocytosis who had a total of 19 pregnancies 8):
- 10 pregnancies occurred in patients before splenectomy, and 9 occurred after splenectomy.
- There were 13 term births, 4 spontaneous abortions (miscarriages), and 2 therapeutic abortions (pregnancy termination for medical indications).
- Of the 19 pregnancies, 8 were complicated by anemia and all were in patients without splenectomy. A hemolytic crisis occurred in 6 pregnancies, and persistent anemia occurred in 2 pregnancies. Transfusion was required in 4 pregnancies 9).
It would appear that pregnancy may cause hemolytic anemia, but maternal morbidity and fetal outcome seem more favorable after splenectomy than before splenectomy 10).
In terms of pregnancy management, folic acid supplementation is necessary. Monitoring for worsening of anemia with complete blood counts and reticulocyte counts is recommended 11).
How might splenectomy affect pregnancy?
Few studies have investigated the effect of splenectomy on pregnancy. Most of the studies performed looked at neonatal outcome among women with immune thrombocytopenia (ITP) who have had a splenectomy, and very few have focused on obstetric outcome.
A study that investigated pregnancy in patients with hereditary spherocytosis (hereditary spherocytosis) found that only about one third of pregnancies in non-splenectomized women developed anemia, or anemia deteriorated. The authors noted that in splenectomized patients, the incidence of complaints was minimal 12).
There has been one retrospective study comparing pregnancies of women who have and have not undergone splenectomy (not specific to women with hereditary spherocytosis). The major finding of this study was that splenectomy is a significant risk factor for adverse obstetric outcomes, and specifically, is an independent risk factor for preterm delivery. However, while there were higher rates of preterm delivery, these deliveries were near term and had no impact on birth outcome 13).
Obstetric complications associated with splenectomy included C-section, maternal blood transfusion, pneumonia during pregnancy, and unspecified complications of anesthesia and sedation during labor. Higher rates of fertility treatments were also found among post-splenectomy women 14).
Importantly, pregnancies following splenectomy were not associated with adverse perinatal outcome – no significant differences were found between the groups regarding low Apgar scores, congenital malformations (birth defects), intrauterine growth restriction (IUGR), or perinatal death 15).
What are the long term effects of removal of spleen and gallbladder in children with hereditary spherocytosis?
Overall, individuals with hereditary spherocytosis who have had their spleen removed showed an improvement in quality of life 16).
Complete removal of the spleen (splenectomy) cures almost all patients with hereditary spherocytosis 17). The spleen, however, is important in fighting infection. Individuals, particularly children, who have had a splenectomy are more likely to contract a serious and possibly life-threatening infection (sepsis). Most septic infections have been observed in children whose spleens were removed in the first years of life, although older children and adults also are susceptible Hereditary Spherocytosis. https://emedicine.medscape.com/article/206107-overview. Subtotal (partial) splenectomy is an effective alternative to total splenectomy; decreasing (but not eliminating) hemolysis (breakdown of red blood cells) and reducing the need for blood transfusions, while maintaining spleen function 18). Subtotal splenectomy, however, is not effective in preventing gallstone formation 19).
Gallbladder removal (cholecystectomy) is a procedure that has been shown to help prevent biliary tract disease and, in some patients with mild hereditary spherocytosis, helps avoid the need for splenectomy. Removal of the gallbladder has not been known to cause any long-term adverse effects, aside from occasional diarrhea.
What are the current recommendations regarding post-splenectomy antibiotic prophylaxis in children?
The ideal duration of antibiotic prophylaxis for children is not clear. Recommendations for daily prophylaxis differ among different authorities 20). Guidelines in the United States suggest relatively limited courses of post-splenectomy prophylaxis (up to five years of age and for at least one year after splenectomy), whereas British guidelines recommend lifelong penicillin prophylaxis in high-risk individuals (defined as those less than 16 or more than 50 years of age, and those with an inadequate response to pneumococcal vaccination) 21).
The American Academy of Pediatrics Committee on Infectious Diseases published a policy statement in 2000 which included the following information 22):
- Antibiotic prophylaxis is recommended for all children with sickle cell disease and functional or anatomic asplenia, regardless of whether they have received pneumococcal immunizations.
- Although the efficacy of penicillin prophylaxis in children with functional or anatomic asplenia other than sickle cell disease has not been studied, it is reasonable to use prophylaxis in the same regimen.
- Antibiotic prophylaxis should be begun before 2 months of age or as soon as sickle cell disease or asplenia occurs or is otherwise recognized or suggested by screening procedures.
- Oral administration of penicillin V potassium is recommended at a dosage of 125 mg twice a day until 3 years of age and at a dosage of 250 mg twice a day after 3 years of age.
- Children who have not experienced invasive pneumococcal infection and have received recommended pneumococcal immunizations may discontinue penicillin prophylaxis after 5 years of age 23).
It has also been stated that individuals in whom prophylaxis is being discontinued should have well-established, regular medical care, and understand the warning symptoms and signs, as well as the management, of possible post-splenectomy sepsis. Individuals with highly compromised immune systems, and survivors of pneumococcal post-splenectomy sepsis, are reasonable candidates for prophylaxis until age 18, or even for life 24).
Individuals looking for specific medical advice for themselves or family members should speak with their health care provider.
Does hereditary spherocytosis increase the risk of stroke or heart attack?
Very rarely, hereditary spherocytosis (hereditary spherocytosis) in people that have not undergone splenectomy has been associated with Moyamoya disease, which can increase the risk of blood clots, strokes, and transient ischemic attacks 25). However, because people who are anemic have lower cholesterol and whole blood viscosity than those who are not anemic, it has been suggested that people with hereditary spherocytosis who have not had their spleen removed should have fewer arteriosclerotic events (such as heart attack or stroke) than unaffected family members. Chronic anemia may slow down the development of arteriosclerosis 26).
People with hereditary spherocytosis who have undergone splenectomy may be at increased risk. Both venous and arterial vascular events have been associated with hereditary spherocytosis in patients who have undergone splenectomy compared to non-splenectomized patients 27). Long-term potential complications of splenectomy in adults may include an increased risk of atherosclerotic heart disease, and many adults who have had a splenectomy are on a low-dose aspirin therapy regimen. More recently, some evidence has pointed to a connection between splenectomy for hereditary spherocytosis and the development of venous thrombosis (blood clots). In some cases, a high platelet count has been suggested as a contributing factor. In others, the connection is less clear. In some patients, pulmonary hypertension has developed related to blood clots in the lung 28).
Hereditary spherocytosis causes
Hereditary spherocytosis is caused by changes (mutations) in at least five genes such as the ANK1, EPB42, SLC4A1, SPTA1, and SPTB genes 29). These genes provide instructions for producing proteins that are found on the membranes of red blood cells. These proteins transport molecules into and out of cells, attach to other proteins, and maintain cell structure. Some of these proteins allow for cell flexibility; red blood cells have to be flexible to travel from the large blood vessels (arteries) to the smaller blood vessels (capillaries). The proteins allow the cell to change shape without breaking when passing through narrow capillaries.
Mutations in red blood cell membrane proteins result in an overly rigid, misshapen cell. Instead of a flattened disc shape, these cells are spherical. Dysfunctional membrane proteins interfere with the cell’s ability to change shape when traveling through the blood vessels. The misshapen red blood cells, called spherocytes, are removed from circulation and taken to the spleen for destruction. Within the spleen, the red blood cells break down (undergo hemolysis). The shortage of red blood cells in circulation and the abundance of cells in the spleen are responsible for the signs and symptoms of hereditary spherocytosis.
Mutations in the ANK1 gene are responsible for approximately half of all cases of hereditary spherocytosis. The other genes associated with hereditary spherocytosis each account for a smaller percentage of cases of this condition.
Inheritance pattern
In about 75 percent of cases, hereditary spherocytosis is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder 30). In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
Less commonly, hereditary spherocytosis is inherited in an autosomal recessive manner 31). This means that to be affected, a person must have a mutation in both copies of the responsible gene in each cell. Affected people inherit one mutated copy of the gene from each parent, who is referred to as a carrier. Carriers of an autosomal recessive condition typically do not have any signs or symptoms (they are unaffected). When 2 carriers of an autosomal recessive condition have children, each child has a:
- 25% (1 in 4) chance to be affected
- 50% (1 in 2) chance to be an unaffected carrier like each parent
- 25% chance to be unaffected and not be a carrier
In some of the cases that result from new mutations in people with no family history of the condition, the inheritance pattern may be unclear 32).
Can my unaffected children pass hereditary spherocytosis on to their children?
The offspring of an individual affected with an autosomal dominant form of hereditary spherocytosis has a 50% (1 in 2) risk to inherit the same mutation in the disease-causing gene. If a child of an affected parent with autosomal dominant hereditary spherocytosis does not inherit the mutation, that child will not pass the mutation on to his/her children because it is not present in his/her genes.
The offspring of an individual with an autosomal recessive form of hereditary spherocytosis will definitely be a carrier of the condition. A carrier of an autosomal recessive condition is generally only at risk to have an affected child if his/her partner is also a carrier for the condition, having a mutation in the same disease-causing gene. When 2 carriers of an autosomal recessive condition have children, each child has a 25% (1 in 4) risk to be affected. If a carrier has a child with an individual who is not a carrier, that child will not be affected.
Hereditary spherocytosis pathophysiology
The following four abnormalities in red blood cell membrane proteins have been identified in hereditary spherocytosis:
- Spectrin deficiency alone
- Combined spectrin and ankyrin deficiency
- Band 3 deficiency
- Protein 4.2 defects
Spectrin deficiency
Spectrin deficiency is the most common defect in hereditary spherocytosis. The biochemical nature and the degree of spectrin deficiency are reported to correlate with the extent of spherocytosis, the degree of abnormality on osmotic fragility test results, and the severity of hemolysis.
Spectrin deficiency can result from impaired synthesis of spectrin or from quantitative or qualitative deficiencies of other proteins that integrate spectrin into the red cell membrane. In the absence of those binding proteins, free spectrin is degraded, leading to spectrin deficiency.
The spectrin protein is a tetramer made up of alpha-beta dimers. Mutations of alpha-spectrin are associated with recessive forms of hereditary spherocytosis, whereas mutations of beta-spectrin occur in autosomal dominant forms of hereditary spherocytosis 33).
Synthesis of alpha-spectrin is threefold greater than that of beta-spectrin. The excess alpha chains normally are degraded. Heterozygotes for alpha-spectrin defects produce sufficient normal alpha-spectrin to balance normal beta-spectrin production. Defects of beta-spectrin are more likely to be expressed in the heterozygous state because synthesis of beta-spectrin is the rate-limiting factor.
Red cell membranes isolated from individuals with autosomal recessive hereditary spherocytosis have only 40-50% of the normal amount of spectrin (relative to band protein 3). In the autosomal dominant form of hereditary spherocytosis, red cell spectrin levels range from 60-80% of normal.
Approximately 50% of patients with severe recessive hereditary spherocytosis have a point mutation at codon (969) that results in an amino acid substitution (alanine [Ala]/aspartic acid [Asp]) at the corresponding site in the alpha-spectrin protein. This leads to a defective binding of spectrin to protein 4.1. Mutations involving the alpha-spectrin beta-spectrin gene also occur, each resulting in spectrin deficiency.
Several other beta-spectrin mutations have been identified. Some of these mutations result in impaired beta-spectrin synthesis. Others produce unstable beta-spectrins or abnormal beta-spectrins that do not bind to ankyrin and undergo proteolytic degradation.
Ankyrin defects
hereditary spherocytosis is described in patients with translocation of chromosome 8 or deletion of the short arm of chromosome 8, where the ankyrin gene is located. Patients with hereditary spherocytosis and deletion of chromosome 8 have a decrease in red cell ankyrin content.
Ankyrin is the principal binding site for spectrin on the red cell membrane. Studies of cytoskeletal protein assembly in reticulocytes indicate that ankyrin deficiency leads to decreased incorporation of spectrin. In hereditary spherocytosis caused by ankyrin deficiency, a proportional decrease in spectrin content occurs, although spectrin synthesis is normal. Of particular interest, 75-80% of patients with autosomal dominant hereditary spherocytosis have combined spectrin and ankyrin deficiency and the two proteins are diminished equally.
Band 3 deficiency
Band 3 deficiency has been recognized in 10-20% of patients with mild-to-moderate autosomal dominant hereditary spherocytosis. These patients also have a proportionate decrease in protein 4.2 content on the erythrocyte membrane. In some individuals with hereditary spherocytosis who are deficient in band 3, the deficiency is considerably greater in older red blood cells. This suggests that band 3 protein is unstable.
Protein 4.2 (pallidin) deficiency
Hereditary hemolytic anemia has been described in patients with a complete deficiency of protein 4.2. Red blood cell morphology in these cases is characterized by spherocytes, elliptocytes, or sphero-ovalocytes.
Deficiency of protein 4.2 in hereditary spherocytosis is relatively common in Japan. One mutation that appears to be common in the Japanese population (resulting in protein 4.2 Nippon) is associated in the homozygous state with a red cell morphology described as spherocytic, ovalocytic, and elliptocytic. Another mutant protein 4.2 (protein 4.2 Lisboa) is caused by a deletion that results in a complete absence of protein 4.2. This is associated with a typical hereditary spherocytosis phenotype.
Aquaporin-1
In addition to abnormal levels of proteins affected by mutations, patients with hereditary spherocytosis may demonstrate aberrant distribution of other proteins in erythrocytes. Crisp et al 34) found reduced expression of the water channel protein aquaporin-1 (AQP1) in the membranes of erythrocytes from patients with hereditary spherocytosis, compared with normal controls. The AQP1 content in erythrocyte membranes correlated with the clinical severity of hereditary spherocytosis.
Red blood cell antibodies
Using a mitogen-stimulated direct antiglobulin test, Zaninoni and colleagues found red blood cell antibodies in 61% of patients with hereditary spherocytosis. Patients with red blood cell-bound IgG of more than 250 ng/mL (the positive threshold of autoimmune hemolytic anemia) had increased numbers of spherocytes and mainly had spectrin deficiency. These researchers concluded that the more evident hemolytic pattern in patients with red blood cell autoantibodies suggests that these antibodies have a pathogenic role in red blood cell opsonization and removal by the spleen 35).
Hereditary spherocytosis symptoms
Clinically, hereditary spherocytosis shows marked heterogeneity, ranging from an asymptomatic condition to fulminant hemolytic anemia. Patients with severe cases may present as neonates, while those with mild hereditary spherocytosis may not come to medical attention until adulthood, when an environmental stressor uncovers their spherocytosis.
The symptoms of hereditary spherocytosis are minor in some children, but for many children the condition is more serious. Your child may get these common symptoms of anemia:
- Pale skin, lips or nail beds compared to their normal color
- Feeling tired or irritable
- Feeling dizzy or lightheaded
- Rapid heartbeat
Your child may also have jaundice (yellow color in the whites of the eyes; maybe yellow tint in the skin for some skin colors). This happens when red blood cells break down and their pigment, called bilirubin, builds up in the body.
The extra bilirubin increases the chance that a newborn may need to be under blue lights (light therapy or phototherapy).
Extra bilirubin also increases the chance of having gallstones.
Hereditary spherocytosis complications
Some individuals with the severe form of hereditary spherocytosis have short stature, delayed sexual development, and skeletal abnormalities.
Many patients with hereditary spherocytosis have bone marrow that is able to compensate enough so that the child is only mildly anemic and does not have major symptoms. Whether your child has mild, moderate or severe hereditary spherocytosis, the major complications of hereditary spherocytosis are aplastic or megaloblastic crisis, hemolytic crisis, and cholecystitis and cholelithiasis 36).
- Aplastic crises. This type of crisis often is associated with viral infections. The bone marrow is suppressed by the viral infection and the number of new red blood cells produced is decreased. The patient’s red blood cells are destroyed at their usual rate and this results in a worsening of the anemia. In this type of crisis the patient with hereditary spherocytosis may quickly develop severe anemia and may require a blood transfusion. Symptoms of an aplastic crisis may include increasing pallor (paleness), decreased energy and decreased appetite.
- Hemolytic crises. This is the most frequent type of crisis that occurs in patients with hereditary spherocytosis. It is caused most often by a viral infection and results when there is a sudden increase in red blood cell destruction. It is rarely severe but will result in worsening anemia, increasing jaundice, enlargement of the spleen and an increased reticulocyte count. Occasionally this type of crisis requires a blood transfusion.
- Gallstones. The excessive production of bilirubin from the destroyed red blood cells can lead to the formation of bilirubin gallstones. These may collect in the bile ducts or gall bladder and cause irritation or obstruction of bile flow. This is called a “gall bladder attack” or cholecystitis. These gallstones may occur in infancy but typically appear in adolescence and young adult life. Five percent of children under 10 with hereditary spherocytosis have gallstones. Studies have shown that approximately 50% of patients with hereditary spherocytosis who are between the ages of 10 and 30 years have gallstones. Over the age of 30 years the incidence continues to rise so that by age 50 almost all patients with hereditary spherocytosis have had gallstones.
- Other health issues. Severe hereditary spherocytosis has been associated with short stature, delayed sexual maturation, changes in the growth of facial bones, gout, leg ulcers and extramedullary hematopoieses. Extramedullary hematopoiesis is the growth of bone marrow tissue in organs of the body outside of the bone marrow. All of these conditions are rare but can be treated by a splenectomy (removal of the spleen.)
Hereditary spherocytosis prognosis
Overall, the long-term outlook (prognosis) for people with hereditary spherocytosis is usually good with treatment 37). However, it may depend on the severity of the condition in each person. Hereditary spherocytosis is often classified as being mild, moderate or severe 38). People with very mild hereditary spherocytosis may not have any signs or symptoms unless an environmental “trigger” causes symptom onset 39). In many cases, no specific therapy is needed other than monitoring for anemia and watching for signs and symptoms 40). Moderately and severely affected people are likely to benefit from splenectomy 41). Most people who undergo splenectomy are able to maintain a normal hemoglobin level 42). However, people with severe hereditary spherocytosis may remain anemic post-splenectomy, and may need blood transfusions during an infection 43).
Information about life expectancy in the medical literature appears to be limited. However, we are not aware of reports that state that life expectancy is known to be significantly shortened in people without other medical problems who are managed appropriately. In all people who undergo splenectomy, there is a lifelong, increased risk of developing a life-threatening infection (sepsis) 44). Although most septic episodes have been observed in children whose spleens were removed in the first years of life, older children and adults also are susceptible. Fortunately, taking certain precautions can reduce this risk and can prevent minor infections from becoming life-threatening 45).
Hereditary spherocytosis diagnosis
To check for spherocytosis, the doctor will:
- Ask about the health of your child and family members
- Examine your child and feel their abdomen to see if their spleen is larger than normal
- Do blood tests to learn more
Here are some of the things the doctor may look for in your child’s blood:
- The level of red blood cells. This shows whether the child has anemia. The test is called a complete blood count, or CBC.
- The percent of immature red blood cells in the blood. These are called reticulocytes. The level is higher in people with spherocytosis.
- The shape of the red blood cells as seen under a microscope. Red blood cells that look round instead of flat are a sign of spherocytosis.
- How much a special chemical binds to the red blood cell membrane. The test is called a hereditary spherocytosis screen.
- Whether the blood contains antibodies that can destroy red blood cells.
- The level of bilirubin.
The classic laboratory features of hereditary spherocytosis include the following 46):
- Mild to moderate anemia
- Reticulocytosis
- Increased mean corpuscular hemoglobin concentration (MCHC)
- Spherocytes on the peripheral blood smear
- Hyperbilirubinemia
- Abnormal results on the incubated osmotic fragility test. Osmotic fragility is the test performed to make the diagnosis of hereditary spherocytosis. The patient’s red blood cells are suspended in a salt solution and their destruction or fragility is measured. If this test is abnormal, a genetic test for the specific mutation associated with hereditary spherocytosis can be performed.
Hereditary spherocytosis treatment
In most patients with hereditary spherocytosis, no specific therapy is needed other than monitoring for anemia and watching for signs and symptoms of an aplastic crisis, a hemolytic crisis, and/or gallstones.
Your doctor will check your child regularly so they receive the right treatment at the right time. Unless your child’s hereditary spherocytosis case is very mild, your doctor will see them at least once a year to check:
- Any symptoms
- Their level of red blood cells
- The size of their spleen
- The risk for gallstones
In some patients who have severe anemia or other complications, a splenectomy (surgical removal of the spleen) is recommended. This can end the destruction of red blood cells, i.e., following a splenectomy, most patients with hereditary spherocytosis will have a normal levels of hemoglobin and bilirubin. Splenectomy also prevents aplastic and hemolytic crises and significantly decreases the risk of gallstones.
However, there are many potential complications to a splenectomy. Patients who have had a splenectomy are at greater risk for very serious bacterial infections. The exact incidence of infection is unknown but more recent studies have shown it to be 1 percent to 2 percent. These infections are more common in children less than five years of age. For this reason, splenectomy for hereditary spherocytosis is usually delayed until after age five. Vaccinations and use of preventive antibiotics decrease this risk but will not absolutely prevent infections.
It is recommended that all patients who are going to have a splenectomy have a hemophilus influenza B, pneumococcus and meningococcus vaccinations prior to the splenectomy. The pneumococcus vaccination should be repeated every five years. Prophylactic antibiotics are recommended for at least three years following splenectomy and may be recommended for a longer period of time. The most important point to remember is that any fever or illness in a child who’s had a splenectomy should be promptly evaluated by a physician.
Other potential complications of a splenectomy include bleeding (during or immediately following the surgery), pancreatitis and/or intestinal obstruction. Long-term potential complications include infection, portal vein thrombosis and intestinal obstruction. Some patients who have had a splenectomy may have a high platelet count. In adult life, this may increase the risk of atherosclerotic heart disease; many adults who have had a splenectomy are on a low-dose aspirin therapy regimen.
More recently, some evidence has pointed to a connection between splenectomy for hereditary spherocytosis and the development of venous thrombosis (blood clots.) In some cases, the high platelet count has been suggested as contributing to the problem of venous thrombosis but, in other patients, that connection is less clear. In some patients, pulmonary hypertension has developed related to blood clots in the lung.
Pulmonary hypertension is an apparently uncommon but potentially fatal complication that has been described anywhere from eight to 50+ years following a splenectomy. It is unknown if there are other factors that may contribute to the development of this complication and at this time there are no specific recommendations for prevention or routine testing for this problem.
Folic acid
A type of vitamin B (folic acid) may help your child’s body produce more red blood cells.
Folic acid used to be standard treatment, but now diets in the United States tend to be rich in folic acid, so most children don’t need to take extra.
Usually it’s only needed by children whose red blood cells break down very quickly. Ask your child’s doctor whether to use a supplement and, if so, how much to give each day.
Therapy for jaundice
Your newborn will need treatment if they have severe jaundice (yellowing of the whites of the eyes or skin). This is caused by a buildup of bilirubin, the pigment from red blood cells. High levels of bilirubin can cause brain damage if untreated.
The usual treatment is to place your baby under blue lights. This is called light therapy or phototherapy.
Blood transfusions
If your child has very low levels of red blood cells, they may need red blood cells from a healthy donor (a blood transfusion). Your child receives the blood through a vein in their arm. This is most likely to be needed when they are 3 to 8 weeks old.
But many children never need a transfusion, or need one only if they get a certain virus (parvovirus) that temporarily stops their body from making red blood cells.
If your child needs a blood transfusion, they can often get care without having to spend a night in the hospital.
Surgery on the spleen
Because red blood cells get destroyed in the spleen, your child’s condition may be helped by removing all or part of their spleen. This surgery is called a splenectomy or partial splenectomy.
Removing part or all of the spleen slows down how fast red blood cells break down. This improves red blood cell levels and reduces the risk of gallstones.
Some children with hereditary spherocytosis never need their spleen removed. It depends on their red blood cell level and other symptoms.
Because the spleen helps fight certain bacterial infections, doctors try to delay this surgery until your child is at least 5 years old. The goal of removing part – rather than all – of the spleen is to leave enough of the spleen tissue to help fight these certain bacterial infections.
In most cases, our surgeons can do laparoscopic surgery. They remove part of the spleen through small incisions with the aid of a tiny camera, rather than through a large cut.
In some children, the part of the spleen that remains grows larger. If that happens, they need another surgery to remove the whole spleen. Some families choose to have the whole spleen removed at the first surgery. Our team can discuss the pros and cons of these approaches and can arrange for you to have a clinic visit with a surgeon to learn more details.
Without a spleen, the risk of infection is higher. After surgery, your doctor will explain how to help avoid infections and what symptoms to watch for. Fever can be an emergency. It is important to keep immunizations up to date after a splenectomy.
Gallstone treatment
Children with spherocytosis have a greater chance of forming gallstones. These are small, stone like objects that form when the liquid in the gallbladder hardens. This liquid is called bile.
Gallstones can cause pain, infection or other problems if they get stuck in the tubes that lead out of the gallbladder.
If your child gets gallstones, they may need surgery to remove their gallbladder. In many patients, doctors do ultrasounds of the abdomen every few years to look for gallstones.
Spherocytosis causes
Spherocytes are most commonly found in immunologically-mediated hemolytic anemias and in hereditary spherocytosis, but the former would have a positive direct Coombs test and the latter would not. The misshapen but otherwise healthy red blood cells are mistaken by the spleen for old or damaged red blood cells and it thus constantly breaks them down, causing a cycle whereby the body destroys its own blood supply (auto-hemolysis). A complete blood count (CBC) may show increased reticulocytes, a sign of increased red blood cell production, and decreased hemoglobin and hematocrit. The term “non-hereditary spherocytosis” is occasionally used, albeit rarely 47).
Causes of spherocytosis can include 48):
- Warm autoimmune hemolytic anemia
- Cold autoimmune hemolytic anemia/paroxysmal cold hemoglobinuria
- Acute and delayed hemolytic transfusion reactions
- ABO hemolytic diseases of newborn/Rh hemolytic disease of newborn
- Hereditary spherocytosis
- Intravenous water infusion or drowning (fresh water)
- Hypophosphatemia
- Bartonellosis
- Snake bite
- Hyposplenism
- Rh-null phenotype
References [ + ]




{kind=link}