Contents
What is hepatoblastoma
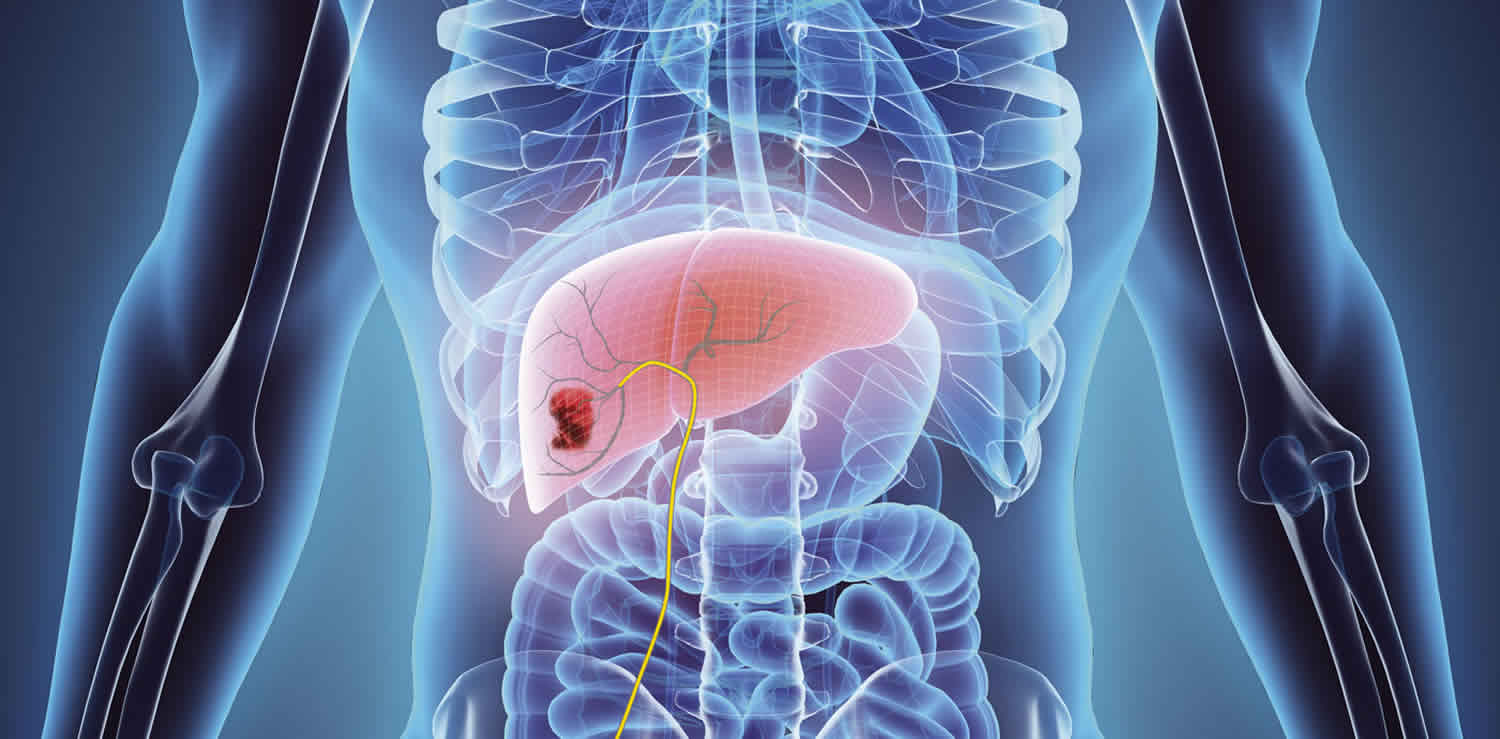
Hepatoblastoma is a very rare type of liver cancer that occurs in infants and children accounting for 80% of malignant liver tumors in childhood 1). Hepatoblastoma typically striking children within the first 3 years of their young lives 2). Hepatoblastoma preferentially affects boys and occurs in infants or very young children at a median age at diagnosis of 16 months 3). Hepatoblastoma incidence is estimated at 1.2–1.5 million children per year, comprising about 1% of all pediatric cancers 4). Over the last 3 decades, advances in chemotherapy and newer surgical techniques have improved survival in patients with localized hepatoblastoma, unfortunately, for the 25% of patients with metastasis (hepatoblastoma stage 4), the overall survival remains poor. The treatment has advanced with neo-adjuvant chemotherapy now the standard of care for most cases. Neo-adjuvant chemotherapy and surgical resection produce a cure rate of approximately 70%, a vast improvement over the dismal 30% cure rate in the 1970s 5). Prognosis is based on many factors including alpha-fetoprotein (AFP) levels, age at the time of diagnosis, completeness of resection, and clinical stage of the disease 6).
What causes hepatoblastoma?
Most cases of hepatoblastoma have unknown cause, but one-third of cases occur in association with specific predisposition syndromes in the setting of normal liver function 7). Hepatoblastoma is strongly associated with premature birth, particularly among low birth weight neonates that weigh < 1,000 g 8). Hepatoblastoma occurs in children from families affected by familial adenomatous polyposis (FAP), which is associated with an inherited germ line mutation of the adenomatous polyposis coli (APC) gene 9). This mutation is also seen in association with Beckwith-Wiedemann syndrome, with which the relative risk of hepatoblastoma is estimated to be 2,280 10). Hepatoblastoma has also been observed in patients with Li-Fraumeni syndrome, Edward syndrome (trisomy 18), nephroblastoma, and Down syndrome (trisomy 21) 11). Evidence has also shown an association with preeclampsia and parental tobacco smoking before and during pregnancy 12). Other factors thought to play a role in pathogenesis include oxygen therapy, certain medication (furosemide), radiation, plasticizers, and total parenteral nutrition (TPN) 13).
The most common genetic mutation involves the Wnt signaling pathway which results in the accumulation of beta-catenin; these mutations are present in a higher proportion of the sporadic cases 14). By immunohistochemistry, beta-catenin usually shows a membranous staining pattern in the more differentiated fetal types and nuclear staining pattern in the less differentiated histologic types 15). In aggressive cases, activation of TERT (human telomerase reverse transcriptase) and MYC signaling has been shown 16).
An American Association for Cancer Research publication suggested that all children with more than a 1% risk of developing hepatoblastoma be screened 17). This includes patients with Beckwith-Wiedemann, hemihyperplasia, Simpson-Golabi-Behmel, and trisomy 18 syndromes. Screening is by abdominal ultrasound and alpha-fetoprotein determination every 3 months from birth (or diagnosis) through the fourth birthday, which will identify 90% to 95% of hepatoblastomas that develop in these children 18).
Risk factors for hepatoblastoma
Conditions associated with an increased risk of hepatoblastoma are described in Table 1.
Table 1. Conditions associated with hepatoblastoma
| Associated Disorder | Clinical Findings |
|---|---|
| Aicardi syndrome 19) | Refer to the Aicardi syndrome section of this summary for more information. |
| Beckwith-Wiedemann syndrome 20) | Refer to the Beckwith-Wiedemann syndrome and hemihyperplasia section of this summary for more information. |
| Familial adenomatous polyposis 21) | Refer to the Familial adenomatous polyposis section of this summary for more information. |
| Glycogen storage diseases I–IV 22) | Symptoms vary by individual disorder. |
| Low-birth-weight infants 23) | Preterm and small-for-gestation-age neonates. |
| Simpson-Golabi-Behmel syndrome 24) | Macroglossia, macrosomia, renal and skeletal abnormalities, and increased risk of Wilms tumor. |
| Trisomy 18, other trisomies 25) | Trisomy 18: Microcephaly and micrognathia, clenched fists with overlapping fingers, and failure to thrive. Most patients (>90%) die in the first year of life. |
Aicardi syndrome
Aicardi syndrome is presumed to be an X-linked condition reported exclusively in females, leading to the hypothesis that a mutated gene on the X chromosome causes lethality in males. The syndrome is classically defined as agenesis of the corpus callosum, chorioretinal lacunae, and infantile spasms, with a characteristic facies. Additional brain, eye, and costovertebral defects are often found 27).
Beckwith-Wiedemann syndrome and hemihyperplasia
The incidence of hepatoblastoma is increased 1,000-fold to 10,000-fold in infants and children with Beckwith-Wiedemann syndrome 28). The risk of hepatoblastoma is also increased in patients with hemihyperplasia, previously termed hemihypertrophy, a condition that results in asymmetry between the right and left side of the body when a body part grows faster than normal 29).
Beckwith-Wiedemann syndrome is most commonly caused by epigenetic changes and is sporadic. The syndrome may also be caused by genetic mutations and be familial. Either mechanism can be associated with an increased incidence of embryonal tumors, including Wilms tumor and hepatoblastoma 30). The expression of both IGFR2 alleles and ensuing increased expression of insulin-like growth factor 2 (IGF-2) has been implicated in the macrosomia and embryonal tumors seen in patients with Beckwith-Wiedemann syndrome 31). When sporadic, the types of embryonal tumors associated with Beckwith-Wiedemann syndrome have frequently also undergone somatic changes in the Beckwith-Wiedemann syndrome locus and IGF-2 32). The genetics of tumors in children with hemihyperplasia have not been clearly defined.
To detect abdominal malignancies at an early stage, all children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia are screened regularly for multiple tumor types by abdominal ultrasonography 33). Screening using alpha-fetoprotein (AFP) levels has also been quite helpful in the early detection of hepatoblastoma in these children 34). Because the hepatoblastomas that are discovered early are small, it has been suggested to minimize the use of adjuvant therapy after surgery 35). However, a careful compilation of published data on 1,370 children with (epi)genotyped Beckwith-Wiedemann syndrome demonstrated that the prevalence of hepatoblastoma was 4.7% in those with Beckwith-Wiedemann syndrome caused by chromosome 11p15 paternal uniparental disomy, less than 1% in the two types of alteration in imprinting control regions, and absent in CDKN1C mutation 36). The authors recommended that only children with Beckwith-Wiedemann syndrome caused by uniparental disomy be screened for hepatoblastoma using abdominal ultrasonography and AFP levels every 3 months from age 3 months to 5 years.
Familial adenomatous polyposis
There is an association between hepatoblastoma and familial adenomatous polyposis (FAP); children in families that carry the APC gene have an 800-fold increased risk of hepatoblastoma. However, hepatoblastoma has been reported to occur in less than 1% of FAP family members, so screening for hepatoblastoma in members of families with FAP using ultrasonography and AFP levels is controversial 37). However, one study of 50 consecutive children with apparent sporadic hepatoblastoma reported that five children (10%) had APC germline mutations 38).
Current evidence cannot rule out the possibility that predisposition to hepatoblastoma may be limited to a specific subset of APC mutations. Another study of children with hepatoblastoma found a predominance of the mutation in the 5′ region of the gene, but some patients had mutations closer to the 3′ region 39). This preliminary study provides some evidence that screening children with hepatoblastoma for APC mutations and colon cancer may be appropriate.
In the absence of APC germline mutations, childhood hepatoblastomas do not have somatic mutations in the APC gene; however, hepatoblastomas frequently have mutations in the beta-catenin gene, the function of which is closely related to APC 40).
Hepatoblastoma histopathology
The cells of hepatoblastoma are similar to fetal liver cells. The histologic types are subdivided into 2 broad categories: epithelial type and mixed type 41). Hepatoblastomas originate from primitive hepatic stem cells that give rise to the epithelial components of the liver. Classically, these tumors are divided into 2 broad categories: epithelial type (E-HB) and mixed epithelial and mesenchymal type (MEM-HB). Revision of this original classification system resulted in the pathology consensus of the pediatric hepatoblastoma classification system, which retained the subdivision of the histologic types into 2 broad categories, as described above. The epithelial type is subdivided into fetal, embryonal, macrotrabecular small cell undifferentiated and cholangioblastic variants, while the mixed type is subdivided into stromal derivatives and teratoid variants.
The fetal subtype is further stratified into 4 categories: well-differentiated; crowded or mitotically active; pleomorphic, poorly differentiated; and anaplastic. The well-differentiated variant is characterized by a low power view demonstrating alternating light and dark areas due to variable cytoplasmic glycogen content. Assessment at higher power reveals a uniform population of hepatocytes arranged in trabeculae that are 2to 3 cells thick. Extramedullary hematopoiesis is a typical finding, and mitotic rate is low. Description of the other variants is beyond the scope of this article.
The embryonal subtype is the most commonly encountered subtype and consists of basophilic cells with scant cytoplasm and increased mitotic rate that is arranged in nests, trabeculae, acini, pseudorosettes, or sheets. The macrotrabecular subtype is arranged in trabeculae that are more than ten cells thick. The SCU subtype consists of dyscohesive, uniform round cells arranged in sheets with increased mitotic activity. Some cases of SCU have a loss of INI1, suggesting a possible association with primary rhabdoid tumors of the liver. The cholangioblastic variant has bile ducts, typically located at the periphery of the epithelial sheets.
The mixed subtype contains a variable combination of epithelial and mesenchymal components. Most commonly, the epithelial component is fetal or embryonal, and the mesenchymal component is osteoid. Stromal derivatives include spindle cells, osteoid, skeletal muscle, and cartilage. Teratoid features include primitive endoderm, neural derivatives, melanin, squamous and glandular elements 42).
Hepatoblastoma symptoms
Hepatoblastomas usually present with as a single, mildly painful, rapidly enlarging abdominal mass that arises in the right lobe of the liver in 55% to 60% of cases 43). Rapid enlargement of these tumors rarely results in tumor rupture and hemorrhage. Hepatoblastoma tumors may reach up to 25 cm in size. Most tumors are solitary; however, up to 15% of tumors are multifocal. Some cases are associated with non-specific symptoms such as weight loss, failure to thrive or anorexia 44). Significant elevations of alpha-fetoprotein (AFP) are observed in 90% of patients, and rarely, a paraneoplastic syndrome can occur.
Hepatoblastoma complications
Hepatoblastoma complications include:
- Intraperitoneal tumor rupture
- Complications related to chemotherapy
- Post-transplant complications
- Psychosocial effects of treatment and painful procedures
Hepatoblastoma diagnosis
Ultrasound and either computed tomography (CT) or magnetic resonance imaging (MRI) are the imaging modalities used to define the extent of tumor involvement of the liver and aid in pre-surgical planning. A chest CT can help detect lung metastasis as up to 20% of cases present with metastases; the lung is the most common location of metastases 45). After imaging, a biopsy, alpha-fetoprotein (AFP) level, liver function tests, and a hepatitis panel are performed as needed.
Biopsy
A biopsy of a pediatric liver tumor is always indicated to secure the diagnosis of a liver tumor, with the exception of the following circumstances:
- Infantile hepatic hemangioma. Biopsy is not indicated in infantile hemangioma of the liver with classic findings on magnetic resonance imaging (MRI). If the diagnosis is in doubt after high-quality imaging, a confirmatory biopsy is done.
- Focal nodular hyperplasia. Biopsy may not be indicated or may be delayed in focal nodular hyperplasia with classic features on MRI using hepatocyte-specific contrast agent. If the diagnosis is in doubt, a confirmatory biopsy is done.
- Children’s Oncology Group (COG) surgical guidelines 46) recommend tumor resection at diagnosis without preoperative chemotherapy in children with PRE-Treatment EXTent of disease (PRETEXT) group I tumors and PRETEXT group II tumors with greater than 1 cm radiographic margin on the vena cava and middle hepatic and portal veins. Therefore, biopsy is not usually recommended in this circumstance.
- Infantile hepatic choriocarcinoma. In infantile hepatic choriocarcinoma, which can be diagnosed by imaging and markedly elevated beta-human chorionic gonadotropin (beta-hCG), chemotherapy without biopsy is often indicated 47).
Tumor markers
The AFP and beta-hCG tumor markers are very helpful in the diagnosis and management of liver tumors. Although AFP is elevated in most children with hepatic malignancy, it is not pathognomonic for a malignant liver tumor 48). The AFP level can be elevated with either a benign tumor or a malignant solid tumor. AFP is very high in neonates and steadily falls after birth. The half-life of AFP is 5 to 7 days, and by age 1 year, it should be less than 10 ng/mL 49).
Hepatoblastoma staging
The Children’s Hepatic Tumors International Collaboration constructed a staging and risk stratification system intended to standardize the assessment of hepatoblastoma cancer across the globe. This new staging system is called the Children’s Hepatic Tumors International Collaboration – Hepatoblastoma Stratification and incorporates confirmed prognostic factors from prior risk stratification systems with new additional factors to stratify patients into 4 risk groups.
Found to be most predictive are alpha-fetoprotein (AFP) levels, patient age, Pretreatment Extent of Disease (PRETEXT) group (I, II, III, or IV), the presence of metastases, and PRETEXT annotation factor. PRETEXT group is based on the extent of the tumor in the liver. PRETEXT annotation factor is determined to be positive if at least 1 of the following 5 factors are present: involvement of the vena cava or all 3 hepatic veins, or both (V); involvement of portal bifurcation or both right and left portal veins, or both (P); extrahepatic contiguous tumor extension (E); multifocal liver tumor (F); tumor rupture at diagnosis (R). Gender, low birth weight, prematurity, and Beckwith-Wiedemann syndrome were not found to be significant. Of note, the histologic type was not included in this risk stratification system but may be incorporated at a future date. Validation of these risk groups is in progress 50).
Table 2. Definitions of PRETEXT/POST-TEXT group (I, II, II, IV) and PRETEXT grouping system annotations V,P,E,M,C,F,N,R.
| PRETEXT/POST-text group | Definition |
|---|---|
| I | One liver section involved |
| Three adjoining sections are tumor free | |
| II | One or two liver sections involved |
| Two adjoining sections are tumor free | |
| III | Two or three liver sections involved |
| One adjoining section is tumor free | |
| IV | Four liver sections involved |
| Annotation: | |
| V | Venous involvement, V, denotes vascular involvement of the retrohepatic vena cava or involvement of ALL THREE major hepatic veins (right, middle, and left) |
| P | Portal involvement, P, denotes vascular involvement of the main portal vein and/or BOTH right and left portal veins |
| E | Extrahepatic involvement of a contiguous structure such as the diaphragm, abdominal wall, stomach, colon, etc. |
| M | Distant metastatic disease (usually lungs, occasionally bone or brain) |
| C | Caudate lobe |
| F | Multifocal tumor nodules |
| N | Lymph node involvement |
| R | Tumor rupture |
Hepatoblastoma prognostic factors
Individual childhood cancer study groups have attempted to define the relative importance of a variety of prognostic factors present at diagnosis and in response to therapy 52). A collaborative group consisting of four study groups (International Childhood Liver Tumors Strategy Group [SIOPEL], Children’s Oncology Group, Gesellschaft für Pädiatrische Onkologie und Hämatologie [GPOH], and Japanese Study Group for Pediatric Liver Tumor [JPLT]), termed Childhood Hepatic tumor International Collaboration (CHIC), have retrospectively combined data from eight clinical trials (N = 1,605) conducted between 1988 and 2010. The CHIC published a univariate analysis of the effect of clinical prognostic factors present at the time of diagnosis on event-free survival (EFS) 53). The analysis confirmed many of the findings described below. The statistically significant adverse factors included the following 54):
- Higher PRETEXT group.
- Positive PRETEXT annotation factors:
- V: Involvement all three hepatic veins and/or intrahepatic inferior vena cava.
- P: Involvement of both left and right portal veins.
- E: Contiguous extrahepatic tumor extensions (e.g., diaphragm, adjacent organs).
- F: Multifocal tumors.
- R: Tumor rupture.
- M: Distant metastases, usually lung.
- Low AFP level (<100 ng/mL or 100–1,000 ng/mL to account for infants with elevated AFP levels) 55).
- Older age. Patients aged 3 to 7 years have a worse outcome in the PRETEXT IV group 56). Patients aged 8 years and older have a worse outcome than do younger patients in all PRETEXT groups.
In contrast, in the SIOPEL-2 and -3 studies, infants younger than 6 months had PRETEXT, annotation factors, and outcomes similar to that of older children undergoing the same treatment 57).
In contrast, sex, prematurity, birth weight, and Beckwith-Wiedemann syndrome had no effect on event-free survival 58).
A multivariate analysis of these prognostic factors has been published to help develop a new risk group classification for hepatoblastoma 59). This classification was used to generate a risk stratification schema to be used in international clinical trials.
Other studies of factors affecting prognosis observed the following:
- PRETEXT group: In SIOPEL studies, having a low PRETEXT group at diagnosis (PRETEXT I, II, and III tumors) is a good prognostic factor, whereas PRETEXT IV is a poor prognostic factor.[42] (Refer to the Tumor Stratification by Imaging and Evans Surgical Staging for Childhood Liver Cancer section of this summary for more information.)
- Tumor stage: In Children’s Oncology Group studies, stage I tumors that were resected at diagnosis and tumors with well-differentiated fetal histology have a good prognosis. These tumors are treated differently than tumors of other stages and histologies 60).
- Treatment-related factors:
- Chemotherapy: Chemotherapy often decreases the size and extent of hepatoblastoma, allowing complete resection.[45-49] Favorable response of the primary tumor to chemotherapy, defined as either a 30% decrease in tumor size by Response Evaluation Criteria In Solid Tumors (RECIST) or 90% or greater decrease in AFP levels, predicted the resectability of the tumor; in turn, this favorable response predicted overall survival among all CHIC risk groups treated with neoadjuvant chemotherapy on the JPLT-2 Japanese national clinical trial.[50][Level of evidence: 2A]
- Surgery: Cure of hepatoblastoma requires gross tumor resection. Hepatoblastoma is most often unifocal and thus, resection may be possible. If a hepatoblastoma is completely removed, most patients survive, but because of vascular or other involvement, less than one-third of patients have lesions amenable to complete resection at diagnosis 61). Thus, it is critically important that a child with probable hepatoblastoma be evaluated by a pediatric surgeon; the surgeon should be experienced in the techniques of extreme liver resection with vascular reconstruction and have access to a liver transplant program. In advanced tumors, surgical treatment of hepatoblastoma is a demanding procedure. Postoperative complications in high-risk patients decrease the rate of overall survival 62). Orthotopic liver transplant is an additional treatment option for patients whose tumor remains unresectable after preoperative chemotherapy 63); however, the presence of microscopic residual tumor at the surgical margin does not preclude a favorable outcome 64). This may be due to the additional courses of chemotherapy that are administered before or after resection 65).
- Tumor marker–related factors: Ninety percent of children with hepatoblastoma and two-thirds of children with hepatocellular carcinoma exhibit the serum tumor marker AFP, which parallels disease activity. The level of AFP at diagnosis and rate of decrease in AFP levels during treatment are compared with the age-adjusted normal range. Lack of a significant decrease in AFP levels with treatment may predict a poor response to therapy 66). Absence of elevated AFP levels at diagnosis (AFP <100 ng/mL) occurs in a small percentage of children with hepatoblastoma and appears to be associated with very poor prognosis, as well as with the small cell undifferentiated variant of hepatoblastoma.[42] Some of these variants do not express INI1 and may be considered rhabdoid tumors of the liver; all small cell undifferentiated hepatoblastomas are tested for loss of INI1 expression by immunohistochemistry 67). Beta-hCG levels may also be elevated in children with hepatoblastoma or hepatocellular carcinoma, which may result in isosexual precocity in boys 68).
- Tumor histology.
Other variables have been suggested as poor prognostic factors, but the relative importance of their prognostic significance has been difficult to define. In the SIOPEL-1 study, a multivariate analysis of prognosis after positive response to chemotherapy showed that only one variable, PRETEXT, predicted OS, while metastasis and PRETEXT predicted event-free survival 69). In an analysis of the intergroup U.S. study from the time of diagnosis, well-differentiated fetal histology, small cell undifferentiated histology, and AFP less than 100 ng/mL were prognostic in a log rank analysis. PRETEXT was prognostic among patients designated group III, but not group IV 70).
Hepatoblastoma treatment
Treatment options for newly diagnosed hepatoblastoma depend on the following:
- Whether the cancer is resectable at diagnosis.
- The tumor histology.
- How the cancer responds to chemotherapy.
- Whether the cancer has metastasized.
Surgical resection is the mainstay of treatment with resectability of the tumor determining the need for neo-adjuvant or adjuvant chemotherapy. At presentation, approximately 60% of tumors are unresectable 71). If unresectable and chemotherapy fails to shrink the tumor to a resectable size, a liver transplant can be done and has a good long-term survival rate 72). The benefit of radiation therapy is unclear, with some unresectable cases responding well. Alpha-fetoprotein levels are useful for tracking surgical success and whether the tumor has metastasized 73). An increased risk of post-transplant lymphoproliferative disorder after immunosuppression for liver transplant has been suggested in some publications 74).
Cisplatin-based chemotherapy has resulted in a survival rate of more than 90% for children with PRETEXT and POST-Treatment EXTent (POSTTEXT) I and II resectable disease before or after chemotherapy 75).
Chemotherapy regimens used in the treatment of hepatoblastoma and their respective outcomes are described in Table 3.
Table 3. Outcomes for Hepatoblastoma Multicenter Trials
| Study | Chemotherapy Regimen | Number of Patients | Outcomes |
|---|---|---|---|
| INT0098 (CCG/POG) 1989–1992 | C5V vs. CDDP/DOXO | Stage I/II: 50 | 4-Year Event-Free Survival/Overall Survival: |
| I/II = 88%/100% vs. 96%/96% | |||
| Stage III: 83 | III = 60%/68% vs. 68%/71% | ||
| Stage IV: 40 | IV = 14%/33% vs. 37%/42% | ||
| P9645 (COG)b 1999–2002 | C5V vs. CDDP/CARBO | Stage I/II: Pending publication | 1-Year Event-Free Survival: |
| I/II: Pending publication | |||
| Stage III: 38 | III/IV: C5V = 51%; CDDP/CARBO = 37% | ||
| Stage IV: 50 | |||
| HB 94 (GPOH) 1994–1997 | I/II: IFOS/CDDP/DOXO | Stage I: 27 | 4-Year Event-Free Survival/Overall Survival: |
| I = 89%/96% | |||
| Stage II: 3 | II = 100%/100% | ||
| III/IV: IFOS/CDDP/DOXO + VP/CARBO | Stage III: 25 | III = 68%/76% | |
| Stage IV: 14 | IV = 21%/36% | ||
| HB 99 (GPOH) 1999–2004 | SR: IPA | SR: 58 | 3-Year Event-Free Survival/Overall Survival: |
| SR = 90%/88% | |||
| HR: CARBO/VP16 | HR: 42 | HR = 52%/55% | |
| SIOPEL-2 1994–1998 | SR: PLADO | PRETEXT I: 6 | 3-Year Event-Free Survival/Overall Survival: |
| SR: 73%/91% | |||
| PRETEXT II: 36 | |||
| PRETEXT III: 25 | |||
| HR: CDDP/CARBO/DOXO | PRETEXT IV: 21 | HR: IV = 48%/61% | |
| Metastases: 25 | HR: metastases = 36%/44% | ||
| SIOPEL-3 1998–2006 | SR: CDDP vs. PLADO | SR: PRETEXT I: 18 | 3-Year Event-Free Survival/Overall Survival: |
| SR: CDDP = 83%/95%; PLADO = 85%/93% | |||
| PRETEXT II: 133 | |||
| PRETEXT III: 104 | |||
| HR: SUPERPLADO | HR: PRETEXT IV: 74 | HR: Overall = 65%/69% | |
| VPE+: 70 | |||
| Metastases: 70 | Metastases = 57%/63% | ||
| AFP <100 ng/mL: 12 | |||
| SIOPEL-4 2005–2009 | HR: Block A: Weekly; CDDP/3 weekly DOXO; Block B: CARBO/DOXO | PRETEXT I: 2 | 3-Year Event-Free Survival/OS: |
| All HR = 76%/83% | |||
| PRETEXT II: 17 | |||
| PRETEXT III: 27 | |||
| PRETEXT IV: 16 | HR: IV = 75%/88% | ||
| Metastases: 39 | HR: Metastases = 77%/79% | ||
| JPLT-1 1991–1999 | I/II: CDDP(30)/THP-DOXO | Stage I: 9 | 5-Year Event-Free Survival/Overall Survival: |
| I = NR/100% | |||
| Stage II: 32 | II = NR/76% | ||
| III/IV: CDDP(60)/THP-DOXO | Stage IIIa: 48 | IIIa = NR/50% | |
| Stage IIIb: 25 | IIIb = NR/64% | ||
| Stage IV: 20 | IV = NR/77% | ||
| JPLT-2 1999–2010 | I: Low-dose CDDP-pirarubicin | PRETEXT I–IV: 212 | 5-Year Event-Free Survival/Overall Survival: |
| I = NR/100% | |||
| II–IV: CITA | II = NR/89% | ||
| III = NR/93% | |||
| IV = NR/63% | |||
| Metastases: High dose chemotherapy + stem cell transplant | Metastases = 32% |
Abbreviations: AFP = alpha-fetoprotein; C5V = cisplatin, 5-fluorouracil (5FU), and vincristine; CARBO = carboplatin; CCG = Children’s Cancer Group; CDDP = cisplatin; CITA = pirarubicin-cisplatin; COG = Children’s Oncology Group; DOXO = doxorubicin; EFS = event-free survival; GPOH = Gesellschaft für Pädiatrische Onkologie und Hämatologie (Society for Paediatric Oncology and Haematology); HR = high risk; IFOS = ifosfamide; IPA = ifosfamide, cisplatin, and doxorubicin; JPLT = Japanese Study Group for Pediatric Liver Tumor; NR = not reported; OS = overall survival; PLADO = cisplatin and doxorubicin; POG = Pediatric Oncology Group; PRETEXT = PRE-Treatment EXTent of disease; SIOPEL = International Childhood Liver Tumors Strategy Group; SR = standard risk; SUPERPLADO = cisplatin, doxorubicin, and carboplatin; THP = tetrahydropyranyl-adriamycin (pirarubicin); VP = vinorelbine and cisplatin; VPE+ = venous, portal, and extrahepatic involvement; VP16 = etoposide.
Footnote: (a) Adapted from Czauderna et al. 76) and Meyers et al. 77); (b) Study closed early because of inferior results in the CDDP/CARBO arm.
Treatment options for hepatoblastoma that is not resectable or not resected at diagnosis
Approximately 70% to 80% of children with hepatoblastoma have tumors that are not resected at diagnosis. Children’s Oncology Group (COG) surgical guidelines recommend biopsy without an attempt to resect the tumor at diagnosis in children with PRETEXT II tumors with less than 1 cm radiographic margin on the vena cava and middle hepatic vein and in all children with PRETEXT III and IV tumors.
Tumor rupture at presentation, resulting in major hemorrhage that can be controlled by transcatheter arterial embolization or partial resection to stabilize the patient, does not preclude a favorable outcome when followed by chemotherapy and definitive surgery 78).
Treatment options for hepatoblastoma that is not resectable or is not resected at diagnosis include the following:
- Chemotherapy followed by reassessment of surgical resectability and complete surgical resection.
- Chemotherapy followed by reassessment of surgical resectability and orthotopic liver transplant 79).
- Transarterial chemoembolization (TACE). TACE may be used to improve resectability before definitive surgical approaches 80).
In recent years, almost all children with hepatoblastoma have been treated with chemotherapy, and in European centers, children with resectable hepatoblastoma are treated with preoperative chemotherapy, which may reduce the incidence of surgical complications at the time of resection 81). Treatment with preoperative chemotherapy has been shown to benefit children with hepatoblastoma. In contrast, an American intergroup study of treatment of children with hepatoblastoma encouraged resection at the time of diagnosis for all tumors amenable to resection without undue risk. The study (COG-P9645) did not treat children with stage I tumors of well-differentiated fetal histology with preoperative or postoperative chemotherapy unless they developed progressive disease 82). In this study, most patients with PRETEXT III and all PRETEXT IV tumors were treated with chemotherapy before resection or transplant.
Patients whose tumors remain unresectable after chemotherapy should be considered for liver transplant 83). In the presence of features predicting unresectability, early coordination with a pediatric liver transplant service is critical 84). In the COG AHEP0731 (NCT00980460) study, early referral (i.e., based on imaging done after the second cycle of chemotherapy) to a liver specialty center with liver transplant capability was recommended for patients with POSTTEXT III tumors with positive V or P and POSTTEXT IV tumors with positive F.
Evidence (chemotherapy followed by reassessment of surgical resectability and complete surgical resection):
- In the SIOPEL-1 study, preoperative chemotherapy (doxorubicin and cisplatin) was given to all children with hepatoblastoma with or without metastases. After chemotherapy, and excluding those who underwent a liver transplant (<5% of patients), complete resection was performed 85).
- The chemotherapy was well tolerated.
- Complete resection was obtained in 87% of children.
- This strategy resulted in an overall survival rate of 75% at 5 years after diagnosis.
- Identical results were seen in a follow-up international study (SIOPEL-2) 86).
- The SIOPEL-3 study compared cisplatin alone with cisplatin and doxorubicin in patients with preoperative standard-risk hepatoblastoma. Standard risk was defined as tumor confined to the liver and not involving more than three sectors 87).
- The rates of resection were similar for the cisplatin (95%) and cisplatin/doxorubicin (93%) groups.
- The overall survival rates were also similar for the cisplatin (95%) and cisplatin/doxorubicin (93%) groups.
- In a pilot study, SIOPEL-3HR, cisplatin alternating with carboplatin/doxorubicin was administered in a dose-intensive fashion to high-risk patients with hepatoblastoma 88).
- In 74 patients with PRETEXT IV tumors, 22 of whom also had metastases, 31 became resectable and 26 underwent transplant. The 3-year overall survival of this group was 69% (± 11%).
- Of the 70 patients with metastases enrolled in the trial, the 3-year event-free survival rate was 56% and the overall survival rate was 62%. Of patients with lung metastases, 50% were able to achieve complete remission of metastases with chemotherapy alone (without lung surgery).
- SIOPEL-4 (NCT00077389) was a multinational feasibility trial of dose-dense cisplatin/doxorubicin chemotherapy and radical surgery for a group of children with high-risk hepatoblastoma. Surgical removal of all remaining tumor lesions after chemotherapy was performed if feasible (including liver transplant and metastasectomy, if needed). Patients whose tumors were resected or whose livers were transplanted after three cycles of chemotherapy subsequently received two postoperative cycles of carboplatin and doxorubicin. Patients whose tumors remained unresectable after three cycles of chemotherapy received two cycles of very intensive carboplatin and doxorubicin before surgery. The primary tumor masses were identified as PRETEXT II (27%), III (44%), and IV (26%) 89).
- Ninety-seven percent of patients (60 of 61) had a partial response with chemotherapy.
- Eighty-five percent of patients (53) underwent complete macroscopic resection; tumor was microscopically present in five patients, all of whom are disease-free survivors.
- Two patients died postoperatively.
- There were 37 partial hepatectomies and 16 liver transplants.
- The study had a total of 62 high-risk patients; 74% of patients (62%–84%) underwent resection. The 3-year disease-free survival (DFS) was 76% and the 3-year overall survival was 83%.
- Of the 16 PRETEXT IV patients, 11 were downstaged after chemotherapy—6 patients to PRETEXT III, 4 patients to PRETEXT II, and 1 patient to PRETEXT I. Twelve tumors became resectable; of these, four patients underwent a partial hepatectomy and eight patients underwent a liver transplant. For patients who presented with PRETEXT IV disease, the 3-year disease-free survival (DFS) was 73% and the 3-year overall survival was 80%.
- In approximately 75% of children and adolescents with initially unresectable hepatoblastoma, tumors can be rendered resectable with cisplatin-based preoperative chemotherapy, and 60% to 65% will survive disease-free 90).
- A combination of ifosfamide, cisplatin, and doxorubicin followed by postinduction resection has also been used in the treatment of advanced-stage disease 91).
In the United States, unresectable tumors have been treated with chemotherapy before resection or transplant 92). On the basis of radiographic imaging, most stage III and IV hepatoblastomas are rendered resectable after two cycles of chemotherapy 93). Some European centers have also used extended resection of selected POSTTEXT III and IV tumors rather than liver transplant 94).
Chemotherapy followed by transarterial chemoembolization (TACE) followed by high-intensity focused ultrasound showed promising results in China for PRETEXT III and IV patients with hepatoblastoma, some of whom were resectable but did not undergo surgical resection because of parent refusal 95).
Treatment options for hepatoblastoma with metastases at diagnosis
The outcomes of patients with metastatic hepatoblastoma at diagnosis are poor, but long-term survival and cure is possible 96). Survival rates at 3 to 5 years range from 20% to 79% 97). To date, the best outcomes for children with metastatic hepatoblastoma resulted from treatment with dose-dense cisplatin and doxorubicin, although significant toxicity was also noted (SIOPEL-4 [NCT00077389] trial) 98).
Treatment options for hepatoblastoma with metastases at diagnosis include the following:
- Chemotherapy followed by reassessment of surgical resectability.
- If the primary tumor and extrahepatic disease (usually pulmonary nodules) are resectable after chemotherapy, surgical resection followed by additional chemotherapy.
- If extrahepatic metastatic disease is in complete remission after chemotherapy and/or surgical resection of lung nodule but the primary tumor remains unresectable, orthotopic liver transplant.
- If extrahepatic metastatic disease is not resectable or the patient is not a transplant candidate, additional chemotherapy, transarterial chemoembolization (TACE), or radiation therapy.
The standard combination chemotherapy regimen in North America is four courses of cisplatin/vincristine/fluorouracil 99) or doxorubicin/cisplatin 100) followed by attempted complete tumor resection. If the tumor is completely removed, two postoperative courses of the same chemotherapy are usually given. Study results for different chemotherapy regimens have been reported.
High-dose chemotherapy with stem cell rescue does not appear to be more effective than standard multiagent chemotherapy 101).
Evidence (chemotherapy to treat metastatic disease at diagnosis):
- A subset of 39 patients presenting with metastases were entered on the SIOPEL-4 (NCT00077389) trial, a multinational feasibility trial of dose-dense cisplatin/doxorubicin chemotherapy and radical surgery for a group of children with high-risk hepatoblastoma. Patients whose tumors were resected or livers transplanted after three cycles of chemotherapy subsequently received two postoperative cycles of carboplatin and doxorubicin. Patients whose tumors were unresectable after three cycles of chemotherapy received two additional cycles of very intensive carboplatin and doxorubicin before surgery 102).
- After three cycles of chemotherapy, there was a complete response (only in the metastases) in 20 of 39 patients and a partial response in 18 of 39 patients. Nineteen of the patients who achieved a complete response were alive without disease 3 years after diagnosis.
- Of the patients who achieved a partial response, seven patients underwent metastasectomy near the time of resection or liver transplant, with an OS of 100%. An additional seven patients with residual small pulmonary nodules underwent resection without metastasectomy; of those, six patients did well and one patient recurred.
- Two patients with initial metastases eventually recurred.
- Liver transplant, rather than resection alone, was needed to treat 7 of the 39 patients who presented with metastases.
- For the subset of 39 patients presenting with metastases, the 3-year disease-free survival was 77% and the overall survival was 79%.
In patients with resected primary tumors, any remaining pulmonary metastasis is surgically removed, if possible 103). A review of patients treated on a U.S. intergroup trial suggested that resection of metastasis may be done at the time of resection of the primary tumor 104).
If extrahepatic disease is in complete remission after chemotherapy, and the primary tumor remains unresectable, an orthotopic liver transplant may be performed 105).
The outcome results are discrepant for patients with lung metastases at diagnosis who undergo orthotopic liver transplant after complete resolution of lung disease in response to pretransplant chemotherapy. Some studies have reported favorable outcomes for these groups 106), while others have noted high rates of hepatoblastoma recurrence 107). All of these studies are limited by small patient numbers; additional study is needed to better define outcomes for this subset of patients. Recent clinical trials have resulted in few pulmonary recurrences in children who underwent liver transplants and presented with metastatic disease 108).
If extrahepatic disease is not resectable after chemotherapy or the patient is not a transplant candidate, alternative treatment approaches include the following:
- Nonstandard chemotherapy agents. Nonstandard chemotherapy agents such as irinotecan, high-dose cisplatin/etoposide, or continuous-infusion doxorubicin have been used 109).
- Hepatic arterial chemoembolization (HACE) or transarterial chemoembolization (TACE) 110).
- Radiation therapy 111).
Treatment options for progressive or recurrent hepatoblastoma
The prognosis for a patient with progressive or recurrent hepatoblastoma depends on several factors, including the following 112):
- Site of recurrence.
- Previous treatment.
- Individual patient considerations.
Treatment options for progressive or recurrent hepatoblastoma include the following:
- Surgical resection. In patients with hepatoblastoma that is completely resected at initial diagnosis, aggressive surgical treatment of isolated pulmonary metastases that develop in the course of the disease, in conjunction with an overall strategy that includes appropriate chemotherapy, may make extended disease-free survival possible 113). If possible, isolated metastases are resected completely in patients whose primary tumor is controlled.[108] A retrospective study of patients in SIOPEL studies 1, 2, and 3 showed a 12% incidence of recurrence after complete remission by imaging and AFP. Outcome after recurrence was best if the tumor was amenable to surgery. Of patients who underwent chemotherapy and surgery, the 3-year Event-Free Survival was 34%, and the Overall Survival was 43% 114). Percutaneous radiofrequency ablation has been used as an alternative to surgical resection of oligometastatic hepatoblastoma 115). Enrollment in a clinical trial should be considered if all of the recurrent disease cannot be surgically removed. Phase I and phase II clinical trials may be appropriate and should be considered.
- Chemotherapy. Analysis of survival after recurrence demonstrated that some patients treated with cisplatin/vincristine/fluorouracil could be salvaged with doxorubicin-containing regimens, but patients treated with doxorubicin/cisplatin could not be salvaged with vincristine/fluorouracil 116). Addition of doxorubicin to vincristine/fluorouracil/cisplatin is under clinical evaluation in the COG study AHEP0731 [NCT00980460]. Combined vincristine/irinotecan and single-agent irinotecan have been used with some success 117). A review of Children’s Oncology Group (COG) phase I and II studies found no promising agents for relapsed hepatoblastoma 118).
- Liver transplant. Liver transplant should be considered for patients with nonmetastatic disease recurrence in the liver that is not amenable to resection 119).
- Percutaneous ablation. Percutaneous ablation techniques may also be considered for palliation 120) or, in some cases, for curative therapy of oligometastatic disease 121).
Hepatoblastoma prognosis
Prognosis is based on numerous factors including age of diagnosis, PRETEXT group, metastases, alfa fetal protein (AFP) levels, histologic subtype, completeness of resection, and clinical stage of the disease. With certainty, the well-differentiated fetal subtype is associated with a better prognosis compared with small cell undifferentiated and macrotrabecular, which are linked to unfavorable prognosis 122). Alfa-fetoprotein is typically high upon diagnosis, but a significant drop after neo-adjuvant chemotherapy portends a better response to treatment. Younger age of diagnosis has historically been a poor prognostic factor; however, recent studies have called this into question, providing evidence that these younger patients do just as well as older children 123). Specifically, children younger than 1 year of age have a better prognosis and children greater than 6 years of age have a worse prognosis 124). Tumor presence at the resection margin, multifocality, and metastases have been shown to be poor prognostic factors. Beta-catenin expression has been shown to be associated with a lower period of event-free survival, while EpCAM expression has been associated with higher tumor viability and a poorer response to neo-adjuvant chemotherapy 125).
Hepatoblastoma survival rate
The 5-year overall survival rate for children with hepatoblastoma is 70% 126). Neonates with hepatoblastoma have outcomes comparable to older children up to age 5 years 127).
References [ + ]




{kind=link}