Contents
Meconium ileus
Meconium ileus is obstruction of the terminal ileum by abnormally thicker and stickier meconium; it most often occurs in neonates with cystic fibrosis. Meconium is the first stool (bowel movement) that a newborn has. This stool is very thick and sticky consisting of succus entericus (bile salts, bile acids, and debris from the intestinal mucosa) and is normally evacuated within 6 hours of birth (or earlier). Meconium ileus accounts for up to 33% of neonatal small-bowel obstructions. Symptoms include vomiting that may be bilious (green), abdominal distention, and failure to pass meconium in the first several days of life. Diagnosis is based on clinical presentation and x-rays. Treatment is enemas with dilute contrast under fluoroscopy and surgery if enemas fail.
Meconium ileus is most often an early manifestation of cystic fibrosis, which causes gastrointestinal secretions to be extremely viscid and adherent to the intestinal mucosa. Meconium ileus is the presenting clinical manifestation of cystic fibrosis in 10 to 20% of cases. Of infants with meconium ileus, 80 to 90% have cystic fibrosis.
Although meconium ileus is usually understood as synonymous with cystic fibrosis until proven otherwise, it may also be seen with pancreatic atresia or stenosis of the pancreatic duct.
Only rarely does meconium ileus occur without cystic fibrosis or pancreatic abnormality, and is thought to be related to gut immaturity (more favourable outcome).
Meconium ileus is classified as either uncomplicated or complicated 1). Uncomplicated meconium ileus is characterized by a distended abdomen often noted at birth and is caused by inspissated meconium in the distal ileum. This is one of the only two causes of neonatal intestinal obstruction that may manifest immediately at birth prior to infant ingestion of air (Figure 1). The other possible cause for such obstruction is in-utero perforation with meconium cyst, which may or may not be caused by meconium ileus. Additional features of the presentation of simple meconium ileus include bilious emesis and the failure to pass meconium. Complicated meconium ileus consists of neonatal bowel obstruction with evidence of necrosis or perforation, and may include intra-abdominal calcification, redness of the abdominal wall, and abdominal tenderness.
Obstruction occurs at the level of the terminal ileum (unlike the colonic obstruction caused by meconium plug syndrome) and may be diagnosed by prenatal ultrasonography. Distal to the obstruction, the colon is narrow and empty or contains small amounts of desiccated meconium pellets. The relatively empty, small-caliber colon is called a microcolon and is secondary to disuse.
In about 50% of meconium ileus cases, complications such as ileal atresia or stenosis, ileal perforation, meconium peritonitis, and malrotation with or without pseudocyst formation can occur in association with meconium ileus 2). The distended loops of small bowel may twist to form a volvulus in utero. If the intestine loses its vascular supply and infarcts, sterile meconium peritonitis can result. The infarcted intestinal loop may be resorbed, leaving an area or areas of intestinal atresia. Infants with meconium ileus are also at increased risk of developing cholestasis.
Figure 1. Meconium ileus
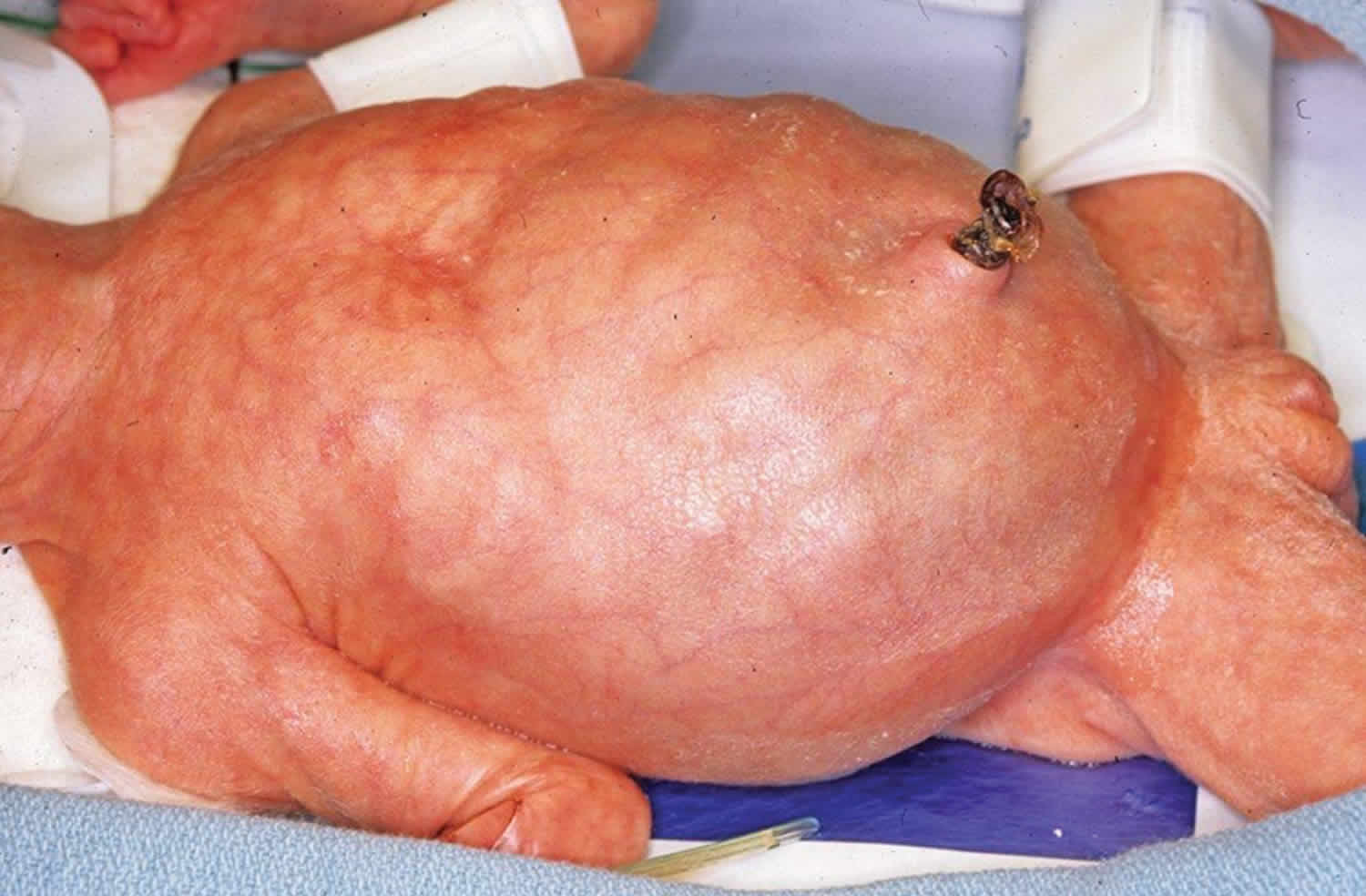
Footnote: Meconium ileus. Newborn with dilated loops of small bowel and a “doughy” abdomen.
[Source 3) ]Meconium ileus causes
Meconium ileus is estimated to occur in 15% of infants with cystic fibrosis. Cystic fibrosis is one of the most common inheritable diseases among Caucasians with an incidence of 1 in 2,500 live births 4). Incidence varies according to race with 93% of cases in the United States occurring among Whites, while only 3.6 and 0.4% cases occur in Blacks and Asian Americans, respectively 5). In the state of California, the overall birth prevalence has been estimated at 19.9 per 100,000 births with 38.8 per 100,000 births for Whites, 37.2 per 100,000 births for Native Americans, and 17.1 per 100,000 births for Blacks 6). Median age of survival for patients with cystic fibrosis has been estimated to be 50 years 7). There does not appear to be a difference in mortality by gender 8).
The pathophysiology of cystic fibrosis is based on an autosomal recessive defect in the gene on the long arm of Chromosome 7. This gene codes for the cystic fibrosis transmembrane conductance regulator (CFTR), a chloride channel on epithelial surfaces. Defects in this receptor cause decreased chloride secretion and increased sodium resorption. Thousands of mutations of the CFTR gene have been identified, but the most common is known as delta F508 9). The CFTR regulates epithelial sodium channels known as ENaCs, so the defect in CFTR results in unregulated ENaCs and increased sodium resorption. In both the pulmonary and gastrointestinal systems, increased sodium resorption is accompanied by water resorption resulting in dehydrated mucus in the lungs and luminal contents of the small bowel 10). Meconium ileus is characterized by intestinal obstruction from the impaction of thickened, protein-rich, meconium inspissated in the distal ileum, possibly related to a deficiency in pancreatic enzymes and abnormal mucin production 11).
Meconium ileus symptoms
After birth, unlike normal neonates, infants with meconium ileus fail to pass meconium in the first 12 to 24 hours. They have signs of intestinal obstruction, including emesis that may be bilious and abdominal distention. Loops of distended small bowel sometimes can be palpated through the abdominal wall and may feel characteristically doughy. Meconium peritonitis with respiratory distress and ascites can occur secondary to perforation.
Simple meconium ileus
Simple meconium ileus is defined as the failure to pass meconium within 48 hours of birth without additional complications 12). Typical management includes an attempted contrast enema and if unsuccessful, celiotomy. Historically, meconium ileus was often treated with an ostomy and postoperative irrigations to break up and evacuate the inspissated meconium. In some cases, these various types of ostomies may still be appropriate. Options for creation of an ostomy include the Bishop-Koop, a distal stoma with a proximal end-to-side anastomosis; Santulli enterostomy, a proximal stoma with a side-to-end distal anastomosis; or Mikulicz, double barrel enterostomy (Figure 3) 13). Current trends in operative management include enterotomy for irrigation with saline or N-acetylcystine. Irrigation may be completed either intraoperatively via the appendix or an enterotomy, or postoperatively through one of the ostomies mentioned previously. Use of a T-tube placed within the intestinal lumen has also been described for postoperative irrigations 14). The T-tube may be removed once the meconium has cleared with spontaneous closure of the fistula 15). Alternatively, an appendicostomy may be created for ongoing irrigations 16). In some cases, a limited bowel resection with primary anastomosis may be necessary after the inspissated meconium has been evacuated.
Complicated meconium ileus
Complicated meconium ileus is characterized by the addition of atresia, volvulus, and perforation, which may result in meconium cyst with peritonitis or gangrene 17). An ultrasound may demonstrate free intraperitoneal fluid with echogenic particles, single or multiple pseudocysts, hepatic or splenic calcifications, and collapsed bowel loops or obstruction 18).
There are four typical presentations of complicated meconium ileus including 19):
- meconium pseudocyst with a calcified fibrous wall and bowel loops peripheral and usually posterior to the cyst,
- dense vascular adhesions with scattered calcifications,
- meconium ascites, and
- infected meconium ascites.
Again, creation of an ostomy is usually necessary to relieve the obstruction and for ongoing irrigation postoperatively. In some of these cases such as meconium cyst, the bowel may be very difficult to find and may be hidden posterior to the cyst rind. To find the bowel, it is necessary to make the abdominal incision laterally and attempt to get behind the cyst rind from the side. Because the bowel may be difficult to discern, an ostomy is usually the best option with later reoperation for establishing intestinal continuity once the inflammation has resolved.
Meconium ileus diagnosis
Diagnosis of meconium ileus is suspected in a neonate with signs of intestinal obstruction, particularly if a family history of cystic fibrosis exists. The earliest signs of meconium ileus are abdominal distention (a swollen belly), bilious (green) vomit and no passage of meconium. Your child’s doctor will order an X-ray of your child’s abdomen to find out if she has meconium in her intestines. A “soap bubble” or “ground glass” appearance due to small air bubbles mixed with the meconium is diagnostic of meconium ileus. If meconium peritonitis is present, calcified meconium flecks may line the peritoneal surfaces and even the scrotum. A water-soluble contrast enema reveals a microcolon with an obstruction in the terminal ileum.
Patients diagnosed with meconium ileus should be tested for cystic fibrosis.
Prenatal ultrasonography can detect changes in utero suggestive of cystic fibrosis and meconium ileus (eg, dilated bowel, polyhydramnios), but these changes are not specific.
Figure 2. Meconium ileus abdominal X-ray
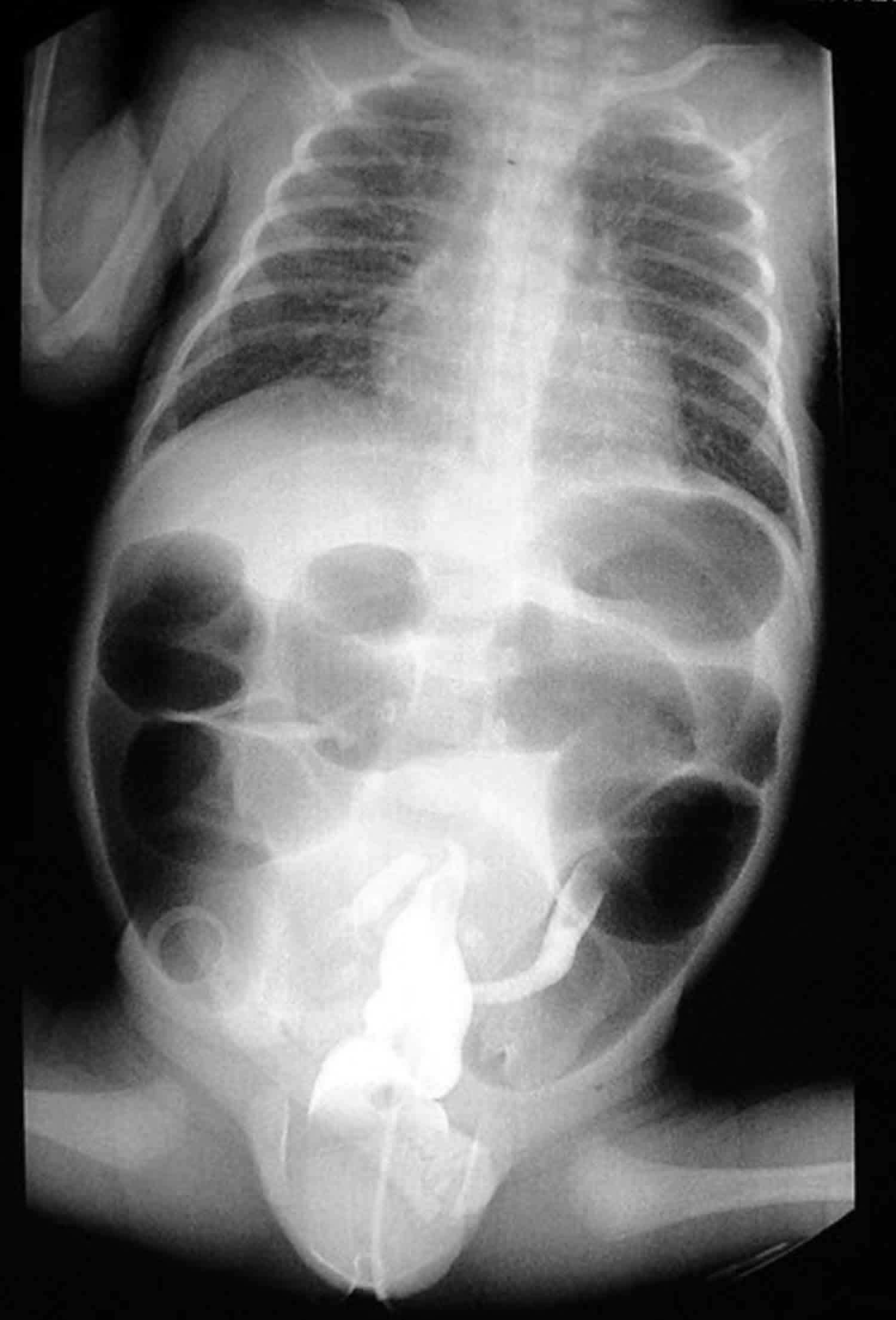
Footnote: Premature child with failure to pass meconium, abdominal distension and vomiting. There is marked dilatation of small bowel loops. Contrast enema reveals small entire colon with radiolucent filling defects representing meconium seen scattered at right colon and distal ileum.
Meconium ileus treatment
If a doctor suspects that your child has a meconium ileus, she won’t be given anything to eat or drink and will be fed through an intravenous (IV) line, a small tube that’s inserted into a vein. A small tube called a nasogastric (NG) tube will also be placed through your child’s nose and passed into her stomach to help remove excess air and fluid.
The medical team may try to break up the meconium blockage with medicines given to your child through an enema. Obstruction may be relieved in uncomplicated cases (ie, without perforation, volvulus, or atresia) by giving ≥ 1 enema with a dilute radiographic contrast medium plus N-acetylcysteine under fluoroscopy; hypertonic contrast material may cause large gastrointestinal water losses requiring IV rehydration.
If the enema does not relieve the obstruction, a laparotomy is required. If your baby needs surgery for meconium ileus, she’ll have a bowel resection and ileostomy placement. A double-barreled ileostomy with repeated N-acetylcysteine lavage of the proximal and distal loops is usually required to liquefy and remove the abnormal meconium.
Nonoperative management
Nonoperative management consists of a hypertonic enema such as Gastrograffin or other contrast enema performed under fluoroscopic guidance. Gastrograffin is a hyperosmolar (1900 mOsm/L), water-soluble, radiopaque solution that contains 0.1% polysorbate 80 and 37% organically bound iodine. 13 14 Prior to initiation of contrast, the patient must undergo fluid resuscitation because the hypertonicity of the enema can lead to significant fluid shifts and cardiovascular collapse in the neonate. It may be acceptable to attempt more than one enema if progress, defined as the passing of contrast more proximally with each enema, is noted on each study. Contrast must be noted passing into the dilated loops of bowel to differentiate meconium ileus from intestinal or colonic atresia. The enema is considered failed if the meconium cannot be evacuated or contrast does not enter the dilated bowel. If a contrast enema is unsuccessful, operative intervention must follow. 13 14
Bowel resection surgery
Your child may need a bowel resection, a surgical procedure that brings part of the small intestine out to the surface of the abdominal wall. This creates an ileostomy, which is temporary. The bowel can be reconnected once your child’s ileus is gone.
Figure 3. Ileostomy
Figure 4. Double-barreled ileostomy
Follow-up care
After the operation, your child will go to the Neonatal Intensive Care Unit (NICU). Your child will have a small incision on her abdomen, which will be covered with a gauze dressing. She’ll also have an NG tube to help empty her stomach. Your child may have an ileostomy. You will learn how to care for the ileostomy before leaving the hospital.
Your baby’s healthcare team will give her pain medication, as needed. When your child first comes back to her room after surgery, she’ll need narcotic medications, such as morphine or Versed, which she’ll get through her IV. After a few days, acetaminophen (paracetamol) should relieve her pain.
Your child will also receive antibiotics after surgery to prevent infection.
Once the bowel has been cleared of meconium, oral feeds with pancreatic enzyme supplementation may be initiated. An evaluation of a cystic fibrosis database from 1990 to 2010 identified 75 neonates with meconium ileus of which 92% underwent laparotomy and 72% received ostomies 20). In this series as well as in others, those who required an ostomy or had complicated meconium ileus had a longer length of stay 21).
Your child will be ready to go home when her incisions are healing nicely, she doesn’t have a fever, and is able to drink, urinate and have a bowel movement. This is usually in one to two weeks.
Once your baby is home, she may drink formula or breast milk. You can give her acetaminophen v — according to her doctor’s instructions — to relieve any pain she’s experiencing. The thin tapes over the incision (called STERI-STRIPS) will fall off on their own.
Your child can have a tub bath one week after surgery.
Be sure to call your child’s doctor if:
- Your child develops a fever greater than 101 degrees F under the arm
- You notice that your child isn’t urinating as often as usual (decreased number of wet diapers)
- Your child is vomiting and unable to eat
- Your child’s surgical incision is swollen or bleeding
Meconium ileus prognosis
Several retrospective cohort studies have examined the association between initial presentation with meconium ileus and later morbidity and mortality. Four retrospective cohort studies that compared pulmonary function and nutritional status for patients diagnosed with cystic fibrosis with meconium ileus versus controls that were diagnosed as a result of other symptoms found no long-term differences between the groups 22). Two earlier studies compared children presenting with meconium ileus with those diagnosed with screening for cystic fibrosis. Both of these studies found diminished pulmonary function and shorter survival for patients with meconium ileus compared with those diagnosed with prenatal screening 23). It is possible that studies comparing patients presenting with meconium ileus and those diagnosed later with symptomatic disease found no difference in long-term outcomes because those with meconium ileus may have more severe disease but benefit from early diagnosis, while those diagnosed later have the disadvantage of delayed diagnosis but the benefit of less severe disease. The study by Lai et al 24) found that those patients with meconium ileus and those diagnosed later with symptomatic disease both had significantly shorter survival than those diagnosed with prenatal or neonatal screening. Alternatively, those patients whose disease was detected with screening may have been biased by less severe phenotypes. Additionally, it appears that the differences may equilibrate with long-term analysis.
Long-term complications
Distal intestinal obstruction syndrome
Previously referred to as meconium ileus equivalent, distal intestinal obstruction syndrome has two presentations: complete and incomplete. Incomplete distal intestinal obstruction syndrome, also known as impending distal intestinal obstruction syndrome, is defined as intestinal obstruction accompanied by a fecal mass in the ileo-cecum and abdominal pain or distension. Complete distal intestinal obstruction syndrome includes all of the previous symptoms with the addition of bilious emesis or air–fluid levels on abdominal radiograph 25). Distal intestinal obstruction syndrome is more prevalent in adults than children with cystic fibrosis and has an estimated prevalence of 15 to 20% of patients with cystic fibrosis 26). Nearly 50% of patients with distal intestinal obstruction syndrome have a history of meconium ileus 27).
The cause of distal intestinal obstruction syndrome is thought to be related to a delayed transit of thickened secretions in the lumen of the gastrointestinal tract 28). It has been proposed that pancreatic insufficiency leads to high fecal fat, which increases stool viscosity and activates the ileal break, delaying transit of intraluminal contents 29). The ileal break is a feedback loop that is activated by fat in the ileum and slows jejunal motility, delays gastric emptying, and reduces small intestinal transit of solid and liquid intraluminal contents 30). This hypothesis has been contradicted by the observation of distal intestinal obstruction syndrome in patients with cystic fibrosis without exocrine pancreatic dysfuntion 31). In the terminal ileum, bile salts normally trigger increased secretion with a mechanism that is dependent on CFTR. A defect in CFTR causes decreased bile acid-stimulated secretion in the terminal ileum, which has been proposed as a part of the mechanism for distal intestinal obstruction syndrome. Malfunction of CFTR also causes increased sodium and water reabsorption through the epithelial sodium channels (ENaCs) in the terminal ileum. 36 Distinct from constipation ,which consists of diffuse stool impaction in the colon, distal intestinal obstruction syndrome is characterized by fecal material causing obstruction at the terminal ileum and in the small bowel 32). Risk factors for distal intestinal obstruction syndrome include a previous history of meconium ileus, pancreatic insufficiency, genotype associated with a severe phenotype, previous history of distal intestinal obstruction syndrome, fat malabsorption, dehydration, and transplantation 33).
Treatment of distal intestinal obstruction syndrome is primarily medical. If the obstruction is incomplete, the patient should receive oral rehydration, stool softeners, and laxatives such as polyethylene glycol 34). If the obstruction is complete without bilious emesis, the patient may be rehydrated and given polyethylene glycol. If the patient presents with complete obstruction and severe bilious emesis, hospitalization with intravenous fluid, nasogastric decompression, and Gastrograffin enemas are required 35). Operative intervention is seldom required with aggressive medical management. However, if the patient fails medical management, celiotomy with enterotomy and possible resection may be necessary 36).
Rectal prolapse
Cystic fibrosis only accounts for approximately 11% of rectal prolapse in pediatric patients; however, 23% of patients with cystic fibrosis will be diagnosed with rectal prolapse 37). Prior to widespread newborn screening, rectal prolapse preceded the diagnosis of cystic fibrosis in more than 40% of cases 38). Since rectal prolapse may be a presenting symptom of undiagnosed cystic fibrosis, testing should be performed in all pediatric patients who present with this finding. 23 40 41 Rectal prolapse has a similar incidence in male and female patients with cystic fibrosis 39). Among patients with cystic fibrosis, those presenting with rectal prolapse have a young average age at diagnosis ranging from 1 to 2.5 years in one series 42 and 3.7 years in another 40). It has been proposed that the higher incidence of rectal prolapse in children younger than 4 years is related to anatomical factors such as the straight course of the rectum, low position of the rectum relative to other pelvic organs, mobility of the sigmoid colon, and weakness of the levator ani muscle 41). In addition, there is only a loose attachment of the mucosa to the underlying muscularis and absence of Houston’s valves in 75% of children under 1 year of age 42). Many of these anatomic factors resolve in early childhood, diminishing the possibility of rectal prolapse. The proposed etiology of rectal prolapse among patients with cystic fibrosis is the presence of large voluminous stool and malnutrition in combination with increased intra-abdominal pressure from chronic coughing 43). It was also noted by Stern et al 44) that of the 29 patients diagnosed with cystic fibrosis who had recurrent rectal prolapse, the problem resolved completely in 72% of patients after pancreatic enzyme supplementation was initiated. In contrast, if patients were already taking supplemental enzymes prior to presentation with rectal prolapse, manipulation of the dosages had a little effect on the rectal prolapse 45).
Typical management of rectal prolapse in children with cystic fibrosis includes manual reduction, which is successful in almost all cases. Occasionally, a patient may experience multiple episodes that are either painful or intolerable 46). In these cases, sclerotherapy, a subcutaneous perianal suture or a sling procedure may be necessary 47). Injection of a sclerosing agent into the rectal submucosa has success rates as high as 90 to 100% 48). A variety of surgical methods have been proposed and the favored technique appears to be institution dependent, and is discussed in greater detail in this edition in the chapter on “rectal prolapse.”
Fibrosing colonopathy
Fibrosing colonopathy is a complication of cystic fibrosis that was first reported in 1994 by Smyth et al and is characterized by concentric rings of fibrosis deep to the submucosa, most frequently in the ascending colon, though it may involve the entire colon. It also involves hypertrophy of the muscularis mucosa as well as inflammation and fibrosis of the submucosa 49). The cause of fibrosing colonopathy was originally thought to be associated with high dosages of enzyme supplementation. This is according to a 1997 study that found a relative risk of fibrosing colonopathy that was double for those on high enzyme doses 50). However, the occurrence of this complication appears to be multifactorial, as there have been cases documented in patients who had never received pancreatic enzyme supplementation 51). It has been hypothesized that increased IgG in response to pancreatic enzymes occurred after ingestion, with a peak elevation at 7 to 9 months following initiation. This timing coincided with the development of fibrosing colonopathy 52). Treatment of fibrosing colonopathy in general includes subtotal versus total colectomy depending on the extent of disease 53). Ostomy may be necessary for rectal strictures though pull-through operations have been described. 29 There are small case reports of using steroids that have allowed strictures to resolve 54).
References [ + ]




{kind=link}