Contents
Juvenile dermatomyositis
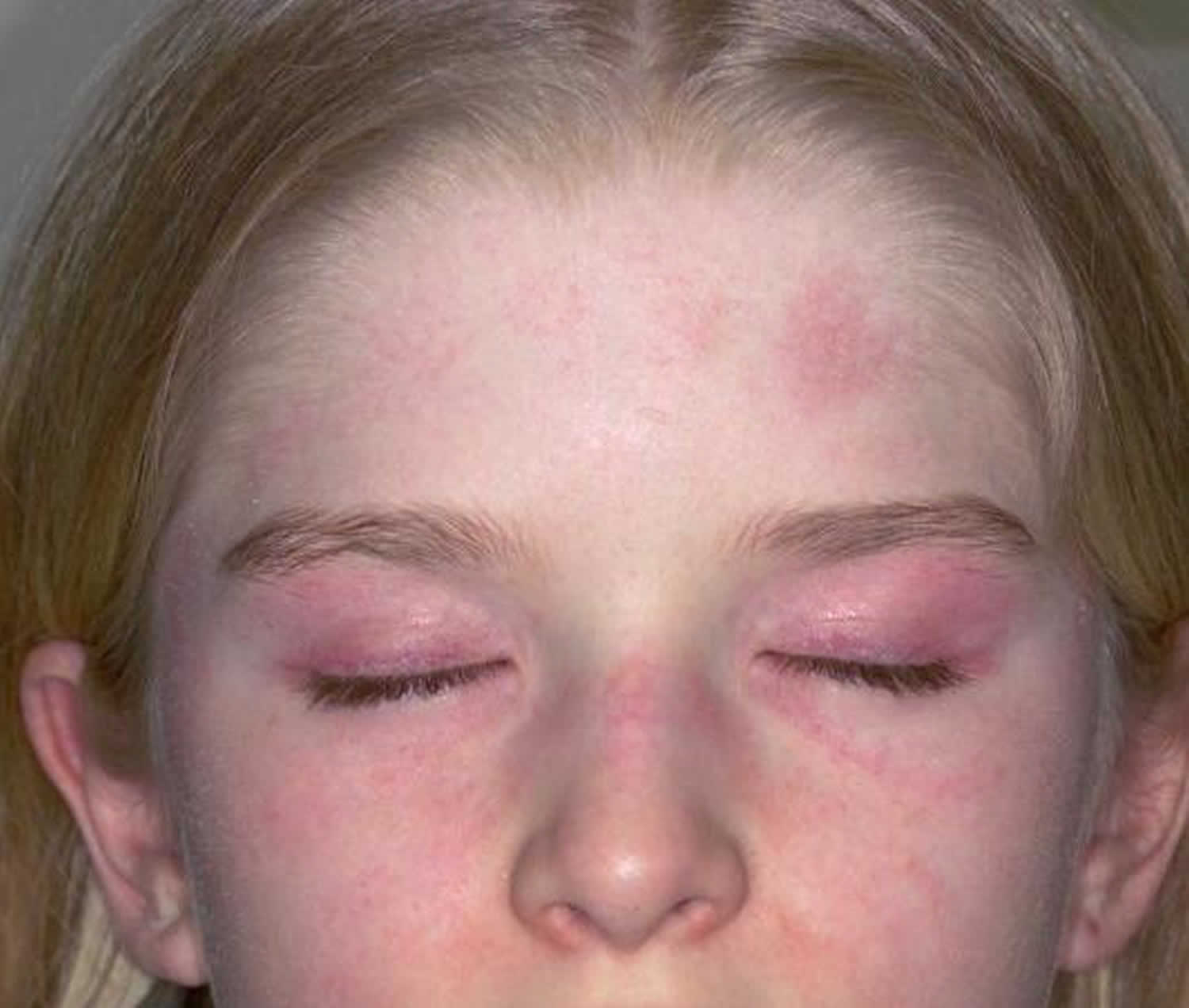
Juvenile dermatomyositis (JDM) is a rare autoimmune inflammatory muscle disorder and vasculopathy that affects children younger than 18 years that causes muscle inflammation (myositis), a skin rash and blood vessels inflammation (vasculitis) that affects about three in one million children each year (3,000-5,000 kids in the United States) 1). Girls are affected about twice as often as boys. Juvenile dermatomyositis is different from other muscle diseases because it also causes skin problems. Symptoms often first appear in children between ages 5 and 10. Children with juvenile dermatomyositis have proximal muscle weakness around the neck, shoulders, and hips. This causes difficulty in climbing stairs, getting into cars, getting up from a chair or off the floor, or brushing hair. Most children have little, if any, pain in their muscles, which distinguishes them from patients with other forms of muscle disease. Many children with other conditions complain of weakness; however, when questioned closely, they really mean that they are tired, short of breath, or depressed rather than suffering from true muscle weakness, as is seen in patients with JDM. They also have a skin rash around certain areas such as the heliotrope rash on the eyelids, knuckles (Gottron papules), and finger joints (see Figure 1 below).
Juvenile dermatomyositis has some similarities to adult dermatomyositis and polymyositis. JDM typically affects children ages 2 to 15 years, with symptoms that include weakness of the muscles close to the trunk of the body, inflammation, edema, muscle pain, fatigue, skin rashes, abdominal pain, fever, and contractures. Children with juvenile dermatomyositis may have difficulty swallowing and breathing, and the heart may also be affected. About 20 to 30 percent of children with juvenile dermatomyositis develop calcium deposits in the soft tissue (calcinosis cutis). Affected children may not show higher than normal levels of the muscle enzyme creatine kinase in their blood but have higher than normal levels of other muscle enzymes 2).
Vasculopathy is considered central to the pathogenesis of juvenile dermatomyositis 3). The exact nature of vasculopathy is not yet understood but it is a complex process with both an inflammatory and a non-inflammatory, occlusive component. Impaired function of JDM vasculature includes immune complex deposition, altered expression of cell adhesion molecules predominantly inducing Th17 cell infiltration, and endothelial cell dysfunction. Development of vasculopathy is associated with the severe extra-muscular manifestations of juvenile dermatomyositis, such as gastrointestinal and cardiac manifestations, interstitial lung disease, ulcerative skin disease or development of calcinosis, and portends a poor prognosis. Correlation of histopathological findings, autoantibodies, and extensive diagnostic workup represent key elements to the early detection of vasculopathic features and early aggressive treatment. Monitoring of vasculopathy remains challenging due to the lack of non-invasive biomarkers.
Juvenile dermatomyositis treatment is aimed at addressing the individual symptoms of each patient. This may involve a combination of medications, physical therapy and supplements 4).
Juvenile dermatomyositis key points
- Juvenile dermatomyositis (JDM) is an inflammatory disease of the muscle (myositis), skin, and blood vessels. Patients with JDM have varying symptoms ranging from mild muscle weakness like difficulty getting out of a chair or difficulty turning over in bed to severe symptoms including profound weakness or difficulty swallowing. Patients can also develop rash or skin changes ranging from mild redness to more severe ulcer formation.
- Other forms of myositis in children include polymyositis, focal myositis, and other rare forms of myositis.
- Myositis almost always causes loss of muscle strength and most all patients also have a rash.
- Early diagnosis and sticking to the treatment plan are important to prevent permanent muscle weakness.
- Children experience JDM differently. While remission is often possible, a minority of children with JDM may have a more chronic course that is less responsive to therapy.
Figure 1. Juvenile dermatomyositis rash
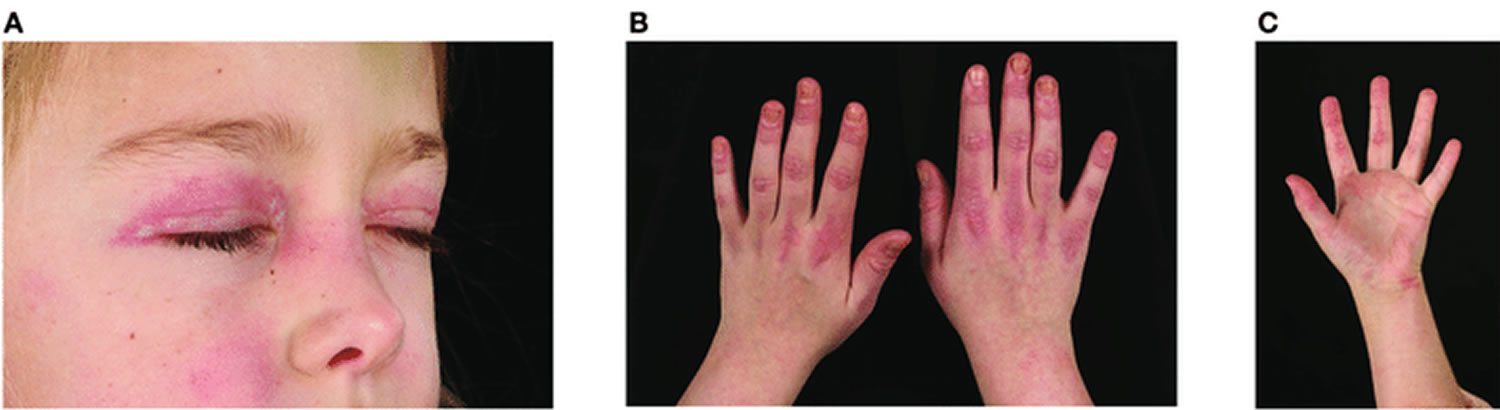
Footnote: Skin changes seen in juvenile dermatomyositis. (A) Heliotrope rash-erythema involving both upper eyelids in a patient with JDM. Written informed parent consent was obtained for the publication of this image. (B) Gottron’s papules over metacarpal and interphalangeal joints with linear extensor erythema. (C) Palmar vasculopathy-palmar erythema, most prominent over the joint creases.
[Source 5) ]Juvenile dermatomyositis causes
The cause of juvenile dermatomyositis is incompletely understood. Dermatomyositis is in a group of diseases or disorders of the muscles called inflammatory myopathies.The leading theory is that the body’s immune system mistakenly directs inflammation against muscle cells and blood vessels in the skin and muscles causing damage, rash, and weakness. Evidence suggests a complex interplay of the innate and adaptive immune systems with environmental triggers in a genetically susceptible host.
Seasonal clustering of juvenile dermatomyositis in the months of April and May suggests the role of environmental triggers in the onset or exacerbation of the disease 6). Infectious agents include viruses, parasites, and bacterial antigens that may produce a break in self-tolerance. Infectious agents implicated include the following 7):
- Coxsackie B virus
- Parvovirus B19
- Enteroviruses
- Streptococcus species
Several mechanisms for infection-triggered autoimmunity have been proposed, including molecular mimicry, induction of anti-idiotypic antibodies, and modification of self-antigens through microbial proteins 8).
Type 1 interferon-alpha/beta genes are overexpressed in dermatomyositis 9). Gene expression profiles of untreated patients may provide indirect evidence of an activated immune response, with an upregulation of interferon alpha/beta genes associated with viral and microbial antigens 10). Type 1 interferons can up-regulate MHC class 1 expression, promote T-cell survival, induce proinflammatory cytokine elaboration and dendritic cell maturation 11).
Levels of type 1 interferon gene expression in peripheral blood mononuclear cells therefore may be a marker for increased disease activity 12). Downregulation of type 1I interferon genes is correlated with clinical improvement in dermatomyositis 13).
Noninfectious agents implicated in the onset of juvenile dermatomyositis include D-penicillamine, vaccinations, and bone marrow transplants 14).
Certain factors that have been associated with adult-onset myositis have not been described in children. These include the following 15):
- Agents related to occupational exposures
- Silica
- Silicone implants
- Lipid-lowering medicines
Patients with human leukocyte antigen DQA1*0501 (HLA-DQA1*0501) have an increased susceptibility to JDM, in a strong linkage disequilibrium to HLA-DR3, compared with age-matched controls in white, black, and Hispanic children 16). The HLA-DQA1*0301 and HLA-DRB*0301 alleles confer an increased risk in whites compared with race-matched controls 17).
Maternally derived chimeric cells have been identified in patients with juvenile dermatomyositis, suggesting a role in pathogenesis. Chimeric cells from mothers with HLA-DQA1*0501 may interact with hosts’ immune responses 18). Microchimerism has been found in 70-100% of muscle tissue and peripheral blood mononuclear cells in patients with juvenile dermatomyositis 19).
Cytokine polymorphisms (eg, the substitution of A to G in the promoter region of tumor necrosis factor [TNF]–alpha-308 allele) is associated with a prolonged, refractory course. The course may be related to an increased production of TNF-alpha in peripheral blood mononuclear cells and muscle fibers of untreated patients with JDM 20). Polymorphisms in the variable number tandem repeat (VNTR) of the interleukin (IL)-1 receptor antagonist have also been implicated as a risk factor in JDM 21).
A study that investigated the association between ultraviolet radiation (UVR) exposure and the clinical and autoantibody expression of juvenile idiopathic inflammatory myopathies found that short-term ultraviolet radiation exposure prior to illness onset may have a role in the clinical and serologic expression of juvenile myositis. Further research examining the mechanisms of action of ultraviolet radiation in the pathogenesis of juvenile idiopathic inflammatory myopathies is needed 22).
Juvenile dermatomyositis symptoms
Inflammation of the blood vessels (vasculitis) causes the primary symptoms of JDM: skin rash and muscle weakness.
The most common signs and symptoms of juvenile dermatomyositis include:
- A violet-colored or dusky-red rash, most commonly on the face, eyelids, and areas around the nails, elbows, knees, chest, and back. The rash, which can be patchy with bluish-purple discolorations, is often the first sign of dermatomyositis. A rash on the knuckles occasionally can be misdiagnosed as eczema when on fact it is dermatomyositis.
- Progressive muscle weakness, particularly in the muscles closest to the trunk (such as those in the hips, thighs, shoulders, upper arms, and neck). This can affect the ability to get out of a chair, off the floor or into the car and leads to falls. The weakness affects both the left and right sides of the body equally, and tends to gradually worsen over time if not treated. Muscle weakness may begin at the same time as the skin rash or develop days, weeks or months after.
Other juvenile dermatomyositis signs and symptoms that may occur include:
- Falling more often.
- Difficulty swallowing (dysphagia)
- Voice changes or weak voice (dysphonia)
- Muscle pain or tenderness
- Weight loss
- Hardened white deposits of calcium under the skin (calcinosis cutis)
- Stomach ulcers and intestinal tears
- Lung problems.
- Feeling very tired or rundown (fatigue).
- Joint pain or stiffness.
- Spiking fever.
Other conditions associated with juvenile dermatomyositis include diabetes, celiac disease, and arthritis.
Juvenile dermatomyositis complications
Complications of juvenile dermatomyositis can happen if the disease is untreated or undertreated.
- Joint contractures. When muscles shorten, either from tissue scarring or in some cases calcium deposits (calcinosis), they can pull joints into a bent position. Daily stretching and physical therapy can prevent permanent damage from joint contractures.
- Skin ulcers. Poor circulation from blood vessel inflammation can cause painful, open sores on the skin.
- Digestive problems. Inflammation of the blood vessels in the intestinal tract can cause ulcers and other digestive issues. Stomach pain or blood in the stool requires an immediate call to the doctor.
Juvenile dermatomyositis diagnosis
There are a number of tests doctors may use to help diagnose juvenile dermatomyositis. These tests include:
- Magnetic resonance imaging (MRI): A scanner creates images of the muscles from data generated by a powerful magnetic field and radio waves. It does not involve any radiation exposure. As MRI has become more sensitive, doctors have been using it more frequently to diagnose myositis. MRI can detect subtle muscle inflammation and swelling early in the disease. A benefit of MRI is that it allows us to view whole muscles to look for patterns or patches of muscle inflammation, instead of taking a small sample from a single muscle.
- Muscle biopsy: Minor surgery is done to remove a small piece of muscle to look at under the microscope. A muscle biopsy may reveal inflammation in the muscles or other problems; it is also helpful to distinguish inflammation from other types of muscle disease such as muscular dystrophy, or infection. In dermatomyositis, inflammatory cells are seen surrounding and damaging the tiny blood vessels within the muscles.
- Blood tests: A blood test will let the doctor know if enzymes from inflamed muscle are elevated. A blood test also can detect specific autoantibodies associated with JDM, which can help in determining the best medication, treatment, and prognosis.
- Laboratory studies in the workup of juvenile dermatomyositis include an erythrocyte sedimentation rate (ESR); muscle enzyme levels; lupus profile (ie, antinuclear antibody [ANA], extractable nuclear antigens [ENA]); and myositis-specific antibody assays such as antibodies against the aminoacyl t-RNA synthetases (ie, anti-Jo-1 antibody), antisignal recognition particle (anti-SRP antibody), and nuclear helicase (anti-Mi-2 antibody) 23).
- Nailfold capillaroscopy: Abnormal swelling and distortion of the blood vessels around the nails can be seen in most patients with JDM 24). This finding suggests active disease. Your doctor can examine the nailbeds by using a lighted magnifying tool.
- Electromyography (EMG) reveals a reduction of the motor unit action potentials in the proximal muscles and fibrillation potentials suggestive of fiber splitting, necrosis, and vacuolization. However, the EMG findings may be normal in approximately 19% of children 25).
Juvenile dermatomyositis treatment
The goal of treatments for juvenile dermatomyositis is to minimize inflammation, improve function, and prevent disability. The treatment should be early and requires a team approach between the rheumatologist, physical therapist, dermatologist, and primary care doctor. juvenile dermatomyositis is unique among most rheumatic diseases of childhood in that it can often be completely cured.
Corticosteroids
Corticosteroids, which are powerful anti-inflammatory drugs, are often used first because they work quickly. Corticosteroids alter the immune system, limiting the production of antibodies and reducing skin and muscle inflammation, as well as improving muscle strength and function. Corticosteroids, especially prednisone, are usually the first choice in treating inflammatory myopathies such as dermatomyositis, because they work quickly. But due to side effects, they are not used for a long time. Once symptoms begin to improve, the doctor will drop the dosage and add or use other medications instead.
The doctor may start with a very high dose, often intravenously (administered directly into vein), and then decrease it as signs and symptoms improve. Signs of improvement may be seen in about two to four weeks as the inflammation is diminished, but full recovery will not be seen for months after initiating treatment. Often, physical therapy is required for strengthening and retraining the muscles that were damaged.
Standard treatment for juvenile dermatomyositis has been high-dose daily oral glucocorticoids (e.g., up to 2 mg/kg/day of prednisone, at times in divided doses), which is continued until clinical and laboratory improvements are evident and then reduced slowly over a two-year period (at least). Intravenous glucocorticoids (methylprednisolone) are also often used at the beginning of therapy. Most patients develop treatment-related side effects with the use of steroids. In many cases, however, prednisone is introduced early as a treatment option and may be discontinued before the two-year period is completed.
Prolonged use of corticosteroids can have serious and wide-ranging side effects, like weight gain, osteoporosis, and cataracts, so the doctor may recommend supplements like calcium and vitamin D to strengthen bone and regular eye exams to detect cataracts.
Corticosteroid-sparing agents
Other medications work slower, but have fewer side effects than corticosteroids like prednisone, and allow the patient to wean off steroids sooner (“spare” the steroids).
- Methotrexate works more slowly than corticosteroids but has fewer side effects. Methotrexate is considered the best initial treatment for most children and is usually started at the same time or right after corticosteroids. It may be given by pill or injection.
- Immunoglobulin contains healthy antibodies from blood donors. They can block harmful antibodies that attack muscle and skin. Immunoglobulin may be administered through an IV (IVIg), because it contains healthy antibodies from blood donors. High doses can block the harmful antibodies that attack muscle and skin.
- Other steroid-sparing agents include cyclosporine, azathioprine, tacrolimus, hydroxychloroquine, mycophenalate mofetil or anti-TNF drugs. and rituximab may be used in very severe disease along with immunoglobulin, steroids, and methotrexate.
Other aspects of treating juvenile dermatomyositis include:
- Skin protection: Protection from ultraviolet A and B (UVA and UVB) light is thought to help control the rash and potentially prevent muscle disease. Use sunscreen or sunblock that decreases exposure to UVA and UVB light. Wear wide-brimmed hats and photo-protective clothing. Avoid sun exposure during peak daylight hours.
- Physical therapy: A physical therapist can teach exercises to maintain and improve strength and flexibility and advise an appropriate level of activity. Physical activity is thought to be important in juvenile dermatomyositis. Physical therapy is directed at preventing muscle wasting and stiffness, and is particularly necessary in patients with calcium deposits (calcinosis) and muscle involvement. Therapy should focus initially on stretching and splinting and only include more aggressive strength-building therapy once inflammation is controlled.
- Speech therapy: If the swallowing muscles are weakened by dermatomyositis, speech therapy can help the patient learn how to compensate for those changes.
- Diet assessment: In juvenile dermatomyositis, chewing and swallowing can become more difficult. A registered dietitian can teach how to prepare foods that are safe to eat.
- Exercise. Regular exercise keeps muscles strong and flexible and prevents muscle weakening and atrophy. Follow an exercise program established by a physical therapist.
Lifestyle and home remedies
While many patients with juvenile dermatomyositis can be cured, some develop a chronic disease; as such, it is important for patients to have good general health practices. These include eating a well-balanced, nutritious diet, maintaining a healthy, weight and managing any other chronic illnesses. Regular exercise is important to regain and maintain strength. It is important for employers, teachers, and family members to understand the limitations imposed by muscle weakness, particularly since patients may look entirely normal.
Stress management
Mind-body techniques, as such meditation and yoga, may help with the psychological and emotional impact of having a chronic illness. Friends, family and trained professionals (such as licensed therapists and psychologists) can also provide support during tough times.
Juvenile dermatomyositis prognosis
Prior to the widespread use of corticosteroids, juvenile dermatomyositis prognosis was poor. One third of patients died from JDM and another third suffered from significant long-term disability 26). Mortality has been related to complications from the vasculopathy, chronic infections, and septicemia 27). Mortality has now declined to 2-3% with improvement in functional outcomes 28).
The average disease duration has varied widely, from 1.5 years to throughout the life span. In general, children with JDM are able to lead normal lives with full recovery, compared with adults 29). Delayed or inadequate treatment with corticosteroids is a predictor of poor outcome and a prolonged disease course 30).
Calcinosis cutis develops in one third of patients and is a major cause of morbidity 31). Calcinosis cutis leads to pain, cosmetic disfigurement, and decreased physical function and quality of life. It may lead to skin atrophy, contractures, nerve entrapment, and ulceration with secondary skin infections 32). Calcinosis has been associated with a delay in diagnosis, lack of aggressive treatment, and cardiac involvement; progression may occur with inadequately treated disease 33). Over one half of patients with JDM develop a chronic disease 24 months after diagnosis; the disease manifests as rash, muscle weakness, or both 34). In severe disease, impairments in physical function may lead to limb contractures.
References [ + ]




{kind=link}